Abstract
Fitzsimmons-Walson-Mellor (F-W-M) syndrome is a rare disease characterized by progressive kidney failure, sensorineural deafness, and variable spastic paraparesis.
We report the case of a 30-year-old male who had been diagnosed with non-progressive connatal encephalopathy, spastic tetraparesis of paraplegic predominance, and intellectual retardation at the age of 14 months. During adolescence, he developed proteinuria, with the subsequent detection of hypertension and elevated plasma creatinine. Analyses revealed a progressive worsening of the glomerular filtration rate, creatinine of 3.56 mg/dL, (CKD-EPI 22 mL/m), non-specific urinary sediment changes, proteinuria (1.8 g/24 h), negativity for antinuclear (ANAs) and antinuclear cytoplasmic (ANCAs) antibodies, normal C3 and C4 complements. Kidney biopsy revealed segmental and focal sclerosis lesions and severe interstitial tubular lesions.
In this complex syndrome, kidney function is affected by focal and segmental sclerotic lesions and interstitial tubule involvement. Differences in the course of kidney function among cases would reflect variations in the intensity of these lesions. Because of the very advanced nature of kidney lesions in the present patient, the biopsy findings did not offer more specific information. However, they appear consistent with the other data obtained. This condition appears to be the first case of F-W-M syndrome detected since its initial description.
Introduction
Fitzsimmons-Walson-Mellor (F-W-M) syndrome is a rare disease characterized by progressive kidney failure, sensorineural deafness, and variable spastic paraparesis. It is named after the authors who first described this syndrome in 1988 [1]. To date, only four cases have been reported of an association between F-M-W syndrome and chronic kidney disease, all in a single family with F-M-W syndrome of probable autosomal dominant heredity; its prevalence is estimated to be lower than 1/1000 000 [2].
Case Presentation
We report the case of a 30-year-old male who had been diagnosed with non-progressive connatal encephalopathy, spastic tetraparesis of paraplegic predominance, and intellectual retardation at the age of 14 months and followed up since then by our neurology department. At the age of 2 years, he had an IQ of 80 and exhibited autistic behavior, and he was diagnosed with bilateral neurosensorial hypoacusia, with hearing loss of 40% in the right ear and 50% in the left. Various fasciotomies were performed to improve his gait. At the age of 8 years, examination by pediatric nephrologists ruled out glomerular and tubular kidney disease, and electrophysiological and muscle biopsy results were normal. During adolescence, he developed proteinuria, with the subsequent detection of hypertension and elevated plasma creatinine. Analyses conducted in the adult nephrology department revealed a progressive worsening of the glomerular filtration rate, creatinine of 3.56 mg/dL, CKD-EPI of 22 mL/m, non-specific urinary sediment changes, proteinuria (1.8 g/24 h), negativity for antinuclear (ANAs)and antinuclear cytoplasmic (ANCAs) antibodies, normal C3 and C4 complements, and negative serology for HIV and hepatitis B and C. Kidney biopsy in February 2016 yielded two cylinders containing a total of 33 glomeruli with focal and segmental hyalinosis/sclerosis lesions, 26 (80%) of which were globally sclerosed. At interstitial level, 50% of tubules were atrophic, with lumina occupied by hyaline cylinders. Myointimal thickening was observed in one artery. Immunofluorescence (IF) study findings were non-specific and showed 10 glomeruli, of which 8 were sclerosed. Electron microscopy (EM) is not available at our center (Figure 1–5). Kidney function continued to progressively decline, and a hemodialysis program was initiated in May 2016.
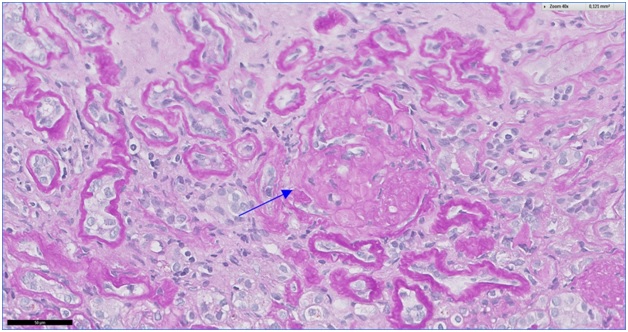
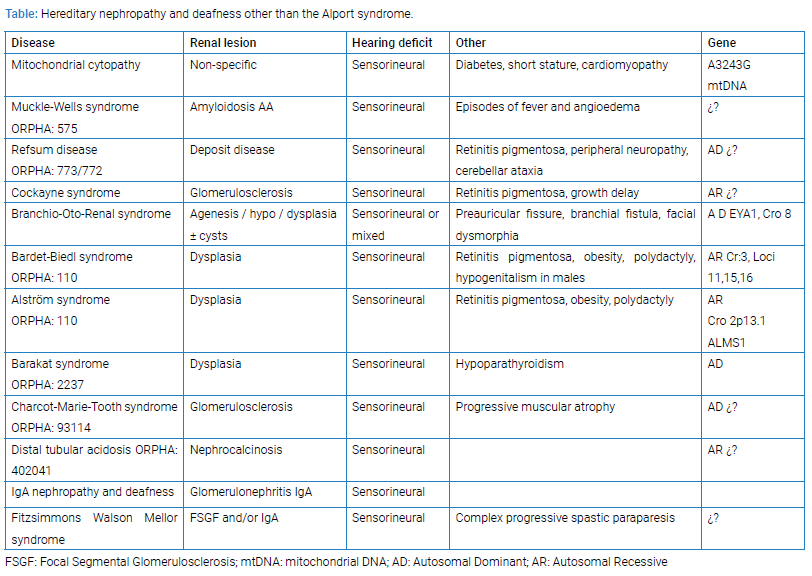
Figure 1: Global glomerulosclerosis is best appreciated in PAS stains (Arrow 40 x).
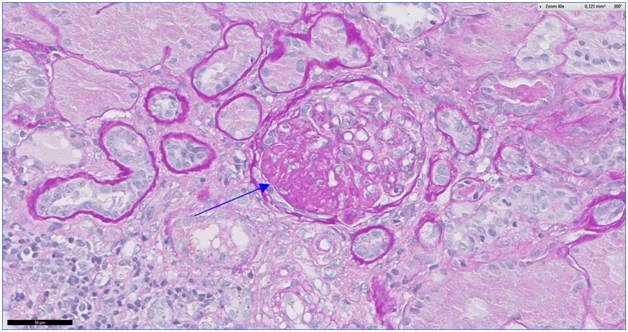
Figure 2: The affected glomerulus shows segmental sclerosing lesion (Arrow PAS 40 x).
The patient received conventional treatment with hemodialysis as well as the treatment prescribed by neurologists. He underwent cadaveric kidney transplantation in April 2017 with a good outcome.
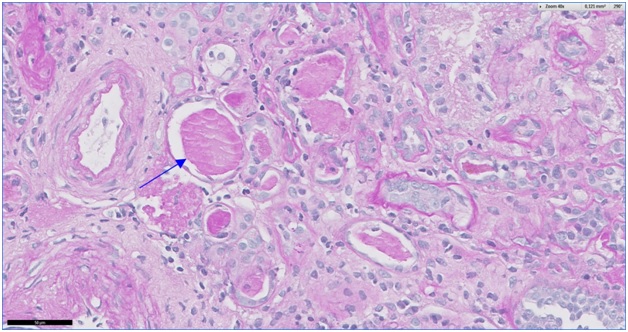
Figure 3: Atrophic and microcystic tubules with eosinophilic casts (Arrow PAS 40 x).
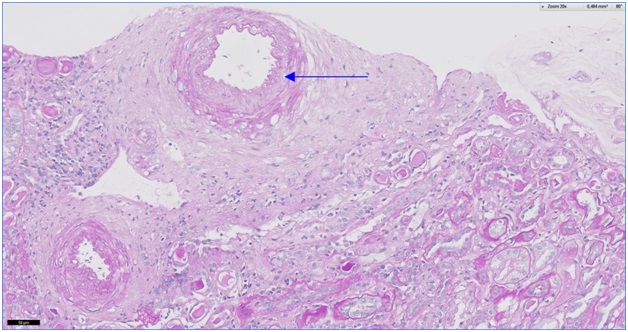
Figure 4: Intimal thickening and fibroelastosis in a small artery (PAS 20 x).
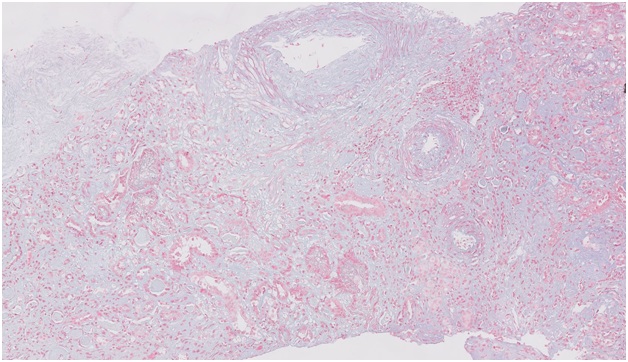
Figure 5: Trichrome stain allows more accurate assessment of interstitial fibrosis; Illustrated here is focal fibrosis with loss of tubules (Masson Trichrome 10 x).
Keywords
Fitzsimmons-Walson-Mellor syndrome; Progressive kidney failure; Sensorineural deafness
Cite this article
Antonio NPC, Maria PO, Juan de Dios FO, Mercedes GM, Mercedes CM. Fitzsimmons-walson-mellor syndrome. Clin Case Rep J. 2020;1(1):1–4.
Copyright
© 2020 Peña Ortega Maria. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).