Similarities and Differences among Genetic Prionopathies: Comparative Description of Familial Creutzfeldt-Jakob Disease and Familial Fatal Insomnia
* Manuel Arias;
Iván Seijo;
Alberto López-Traba;
Alejandro Rivero de Aguilar;
Emilio Rodríguez-Castro;
Susana Arias-Rivas;
Purificación Cacabelos;
-
* Manuel Arias: Department of Neurology, Complexo Hospitalario Universitario, Galicia, Spain.
-
Iván Seijo: Department of Neurology, Complexo Hospitalario Universitario, Galicia, Spain.
-
Alberto López-Traba: Department of Neurology, Complexo Hospitalario Universitario, Galicia, Spain.
-
Alejandro Rivero de Aguilar: Department of Neurology, Complexo Hospitalario Universitario, Galicia, Spain.
-
Emilio Rodríguez-Castro: Department of Neurology, Complexo Hospitalario Universitario, Galicia, Spain.
-
Susana Arias-Rivas: Department of Neurology, Complexo Hospitalario Universitario, Galicia, Spain.
-
Purificación Cacabelos: Department of Neurology, Complexo Hospitalario Universitario, Galicia, Spain.
Abstract
Human prionopathies can be sporadic, hereditary, or acquired disorders. Familial Creutzfeldt-Jakob (F-CJD), Familial Fatal Insomnia (FFI), and Gerstmann-Sträussler-Scheinker (GSS) are the main hereditary variants. We describe the first FFI case in Galicia and other F-CJD, underlying their similitudes and differences.
Case 1 presented with insomnia, behavioral changes, and weight loss. He eventually deceased in 8 months. MRI DWI/FLAIR hyper intensities in lymbic lobes and lenticular nuclei. The periodic pattern in EEG. E200K (129MM) in the PRNP gene.
Case 2 presented with insomnia, pseudosonambulism, and marked weight loss, followed by cognitive decline, diplopia, and ataxia. He passed away in 18 months. MRI: subcortical atrophy. EEG: no periodic pattern. Polysomnography: disorganized NREM sleep. PET-18FDG: thalamic hypometabolism. Genetic study: D178N (129MM) in the PRNP gene.
The onset of sleep disorder and weight loss was similar in both cases. The results in EEG, PSG, MRI, and PET, were clearly different in them.
Introduction
Prion diseases are degenerative diseases of the central nervous system caused by the intracellular accumulation of an abnormal isoform (folded beta, PrPsc) of the so-called as prion protein, whose normal isoform is alfa-hellix (PrPc) [1,2]. PRNP gene (chromosome 20) codifies its synthesis. Prionopathies can be sporadic, inherited or transmitted by accident or iatrogenic [2]. The main hereditary prion diseases are Familial Creutzfeldt-Jakob Disease (F-CJD) and Familial Fatal Insomnia (FFI) [3].
Clinical Cases
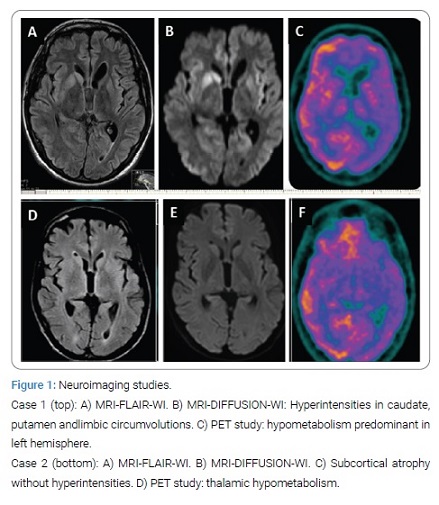
Case 1: A 44-year-old male without relevant past medical history (his father died at middle age from an unknown cause), presented a nonspecific febrile syndrome and thereafter began to lose weight (8 kg in 2 months), and associated progressive insomnia. Three months later, he suffered from balance problems, falls and epileptic seizures with impaired awareness. He was admitted to a hospital, where several studies were performed (serum analysis including paraneoplastic biomarkers, body CT scan, EEG, brain MRI, and CSF studies). Treatment with anti-epileptics, corticosteroids, acyclovir and IGIV were applied. As his condition worsened, he was transferred to our department. Multifocal myoclonus was noted on clinical examination. MRI and CSF studies were repeated. Brain PET-18FDG scan and genetic testing were performed. An E200K heterozygous mutation and a 129MM polymorphism were found in the PRNP gene. The patient worsened and died 8 months after onset. The test results are detailed on (Table 1). (Figure 1) shows the neuroimaging studies.
Case 2: A 48-year-old man started having difficulty in falling asleep. He became unable to sleep on many nights and had somnambulism behavior. At the same time he lost 25 kg in 6 months. He complained about imbalance, tremor, intermittent diplopia, memory loss and difficulties in concentration. In other hospital several tests were carried out: serum analysis with immunological and paraneoplastic markers, body CT scans, intestinal biopsy, Whipple disease tests and brain MRI. No significant abnormalities were found in these studies. A DAT-scan showed asymmetric nigrostriatal degeneration. As he presented occasional visual hallucinations he was diagnosed with Lewy Body Dementia (LBD). He was admitted in our center and he underwent EEG (normal), polysomnography (PSG), CSF studies, brain MRI and PET imaging, and finally PRNP gene analysis, in which D178N, 129MM polymorphisms were found. Results are detailed on (Table 1) and (Figure 1). He died upon 16 months of the symptoms onset.

Discussion
The two described patients are the first genetic prionpathies diagnosed in our department. Case 2 is the first FFI diagnosis in Galicia (Spain); a few clusters were identified in Euskadi and Andalusia [4]. The incidence of human prion diseases is 1–2 per million. F-CJD accounts for 15% of total cases. There are no clinical differences between sporadic and genetic forms, although 129MM polymorphism usually courses with insomnia, dysautonomia and sleep disorganisation, which confers similarities with FFI [5]. About 40 mutations causing F-CJD have been described. E200K is the most common; it has been reported in several countries (Slovakia, Lybia, Tunisia, Italy, Spain, Chile and Argentina): it is believed to have been spread by the Sephardic jewish diaspora after being expelled out of Spain by the Catholic Kings [6,7].
The diagnosis of sporadic CJD is sustained by a clinical picture of rapidly progressive dementia and myoclonus, supported by at least two of the following findings: elevated 14.3.3 protein in CSF [8], periodic pattern in EEG and cortical and basal ganglia hyperintense lesions in MRI-DWI [9,10]. The real-time quaking-induced conversion (RT-QuIC) assays have made an impact on diagnosis of atypical cases [11]. Histopathologic examination provides the definitive confirmation. In genetic forms, molecular analysis is confirmatory. Differential diagnosis includes LBD, Whipple disease, autoimmune/paraneoplastic encephalitis, Hashimoto or Wernicke encephalopathies, and bismuth intoxication.
FFI was first described by Lugaresi et al. in 1986 [12]. It is the result of D178N mutation in the PRNP gene associated with 129 MM or 129MV polymorphisms [13]. Other polymorphisms in the 129 position produce an CJD phenotype. The age of onset is variable; the median survival is 18 months, double as F-CJD. The usual picture consists of an alteration in sleep-wake cycle with sleep fragmentation, accompanied by agitation (Agrypnia agitata), dysautonomia and weight loss, followed by cognitive decline, ataxia and other motor involvement, sometimes with myoclonus. Periodic pattern is not observed in EEG. From the first months a NREM sleep disorganization can be demonstrated in PSG [14]. Thalamic hypomethabolism in FDG-PET stands out as well [15], even in presymptomatic cases, which corresponds with molecular, biochemical and histopathologic findings of thalamic neuronal loss with minimal spongiosis. When comparing neuroimaging studies (Figure 1), MRI findings are markedly different, given that in FFI there are no hyperintense signals in cortical gyri or basal ganglia.
In summary, both familial prion diseases (F-CJD and FFI) manifest initially with sleep disorder and weight loss, more notably in FFI. Evolution is shorter in F-CJD, which presents characteristic findings in EEG and MRI. The contribution of PET stands out by showing thalamic hypomethabolism in FFI. Finally, genetic testing is of primordial value for both diagnostic purposes as well as to provide genetic counseling.
Authors Contributions
All authors participate in the diagnosis and treatment of both patients and also read and approved the final version of paper, planed by MA and wrote by IS, ER-C and MA.
References
- Prusiner SB. Shattuck lecture– –neurodegenerative diseases and prions. N Engl J Med. 2001;344(20):1516–1526.
- Imran M, Mahmood S. An overview of human prion diseases. Virol J. 2011;8:559.
- Mead S. Prion disease genetics. Eur J Hum Genet. 2006;14(3):273–281.
- Zarranz JJ, Digon A, Atarés B, Arteagoitia JM, Carrera N, Fernández-Manchola I, et al. Familial prion diseases in the Basque Country (Spain). 2005;24(1– 2):103–109.
- Goldfarb LG, Petersen RB, Tabaton M, Brown P, Le Blanc AC, Montagna P, et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism. Science. 1992;258(5083):806–808.
- Goldfarb LG, Korczyn AD, Brown P, Chapman J, Gajdusek DC. Mutation in codon 200 of scrapie amyloid precursor gene linked to Creutzfeldt–Jakob disease in Sephardic Jews of Libyan and non-Libyan origin. Lancet. 1990;336:637–638.
- Chapman J, Brown P, Goldfarb LG, Arlazoroff A, Gajdusek DC, Korczyn AD. Clinical heterogeneity and unusual presentations of Creutzfeldt–Jakob disease in Jewish patients with the PRNP codon 200 mutation. J Neurol Neurosurg Psychiatry. 1993;56(10):1109–1112.
- Muayqil T, Gronseth G, Camicioli R. Evidence-based guideline: diagnostic accuracy of CSF 14-3-3 protein in sporadic Creutzfeldt-Jakob disease: report of the guideline development subcommittee of the American Academy of Neurology. Neurology. 2012;79(14):1499–1506.
- Vitali P, Maccagnano E, Caverzasi E, Henry RG, Haman A, Torres-Chae C, et al. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology. 2011;76(20):1711–1719.
- Wang LH, Bucelli RC, Patrick E, Rajderkar D, Alvarez Iii E, Lim MM, et al. Role of magnetic resonance imaging, cerebrospinal fluid, and electroencephalogram in diagnosis of sporadic Creutzfeldt-Jakob disease. J Neurol. 2013;260(2):498–506.
- Green AJE. RT-QuIC: a new test for sporadic CJD. Pract Neurol. 2019;19(1):49–55.
- Lugaresi E, Medori R, Montagna P, Baruzzi A, Cortelli P, Lugaresi A, et al. Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei. N Engl J Med. 1986;315(16):997–1003.
- Montagna P, Cortelli P, Avoni P, Tinuper P, Plazzi G, Gallassi R, et al. Clinical features of fatal familial insomnia: phenotypic variability in relation to a polymorphism at codon 129 of the prion protein gene. Brain Pathol. 1998;8(3):515–520.
- Sforza E, Montagna P, Tinuper P, Cortelli P, Avoni P, Ferrillo F, et al. Sleep-wake cycle abnormalities in fatal familial insomnia. Evidence of the role of the thalamus in sleep regulation. Electroencephalogr Clin Neurophysiol. 1995;94(6):398–405.
- Prieto E, Dominguez-Prado I, Riverol M, Ortega-Cubero S, Ribelles MJ, Luquin MR, et al. Metabolic patterns in prion diseases: an FDG PET voxel-based analysis. Eur J Nucl Med Mol Imaging. 2015;42(10):1522–1529.
Keywords
Prionopathies; Human prion disease; Familial fatal insomnia; Familial Creutzfeldt-Jakob disease
Conflict of Interest
The authors claim no conflicts of interest.
Cite this article
Arias M, Seijo I, López-Traba A, de Aguilar AR, Rodríguez-Castro E, Arias-Rivas S, et al. Similarities and differences among genetic prionopathies: comparative description of familial Creutzfeldt-Jakob disease and familial fatal insomnia. Clin Case Rep J. 2020;1(5):1–3.
Copyright
© 2020 Manuel Arias, This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
