Abstract
COVID-19 called Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) is an ongoing outbreak that has caused a crucial global public health issue over many countries. The incidence rate is significantly increased in the entire world, including Korea. In this study, we conducted an in-depth analysis of mutations using 126 sequences available in public. In total, 177 mutation sites were detected using multiple sequences alignment. We observed that the only 30 mutations were shared with more than two other individual’s virus genome, and 13 mutations of them originated from China and Wuhan. It highlighted that there are specific mutations only exhibited in Korean samples, and they correspond to a number of multi-individual variants that indicate a distinct pattern compared to the mutations of other countries. This research could support developing the targeted therapy of infection and regarded as the aid of further epidemiological investigation.
Introduction
COVID-19 has evoked an emergency of international concerns as the number of confirmed cases has globally exceeded 100,000 in the world as of March 7, 2020 [1]. Although the COVID-19 is considered less virulent, it is likely to transmit more readily among humans than SARS [2]. When it comes to severe risk of the COVID-19, it has spread to many countries, resulting in more than deaths of 116,000 and 1.8 million infected people [3].
Betacoronavirus, a genus that consists of the SARS-CoV-2, has a certainty of characteristics such as being a single-stranded positive-sense RNA, and a high likelihood of having hosted in mammals particularly [4].
Some researchers have argued that the COVID-19 identified as subgenus of Sarbecovirus of Betacoronavirus through phylogenetic analysis to be interpreted differently with SARS-CoV and MERS-CoV (Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. 2020). Although the 2019-nCoV is known to possess a high potential of severe illness in many patients, it initially did not seem to transmit between people as rapidly as it does as for now [5]. However, more recent epidemiological data insist that it is more likely to suffer the human host adaptation and to contain a speedy spread of human to human transmission [6,7].
As the WHO-China Joint Mission emphasized the importance of the transmission dynamics during the epidemic growth phase, some researchers have insisted that considering the Hua Nan market as the origin of SARS-CoV-2 was not rational but admitted it as the only source caused SARS-CoV-2 transmission to humans [8]. In a state of scarce information on potential intermediary origins, the demand of seeking the origin and transmission pattern of SARS-CoV-2 is increased. In the aftermath of the infection rates of SARS-CoV-2 largely expanded across China, the confirmed cases outside China have been increased correspondingly. The outbreak and transmission source needs to be considered as important to establish strategies in terms of prevention, including infection. We could infer the path of virus transmission and identify novel mutations related to the transmission by exhibiting the evolutionary history driven by different SARS-CoV-2 samples.
In this study, we outline phylogenetic analyses based on full-length COVID-19 genomes from GISAID. We demonstrate that the phylogenetic approach would support our study with epidemiological studies to investigate the source of SARS-CoV-2 and the direction of human-to-human transmissions as providing relevant evidence. We also determine to infer the transmission history and evolutionary relationships of the worldwide samples by characterizing genomic variations of SARS-CoV-2.
Materials and Methods
Virus genome: In total, 126 virus genome sequences were obtained from the GISAID database (access date February 12, 2020) to decode the evolution and transmissions of SARS-CoV-2 in the recent two months [9] (Table 1).
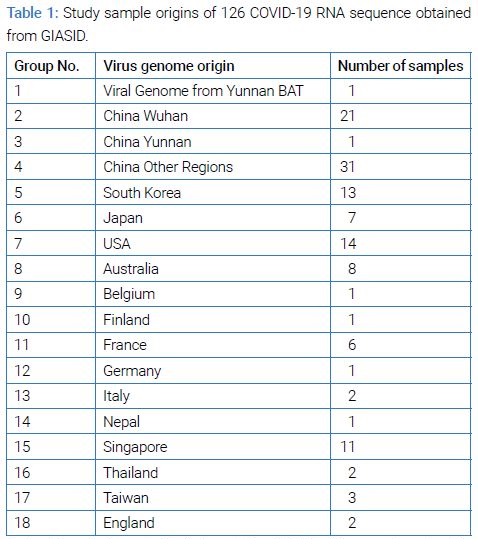
With a total of the virus genome sequences, the most significant number of countries from 53 samples (21 originated from Wuhan, one from Yunnan, 31 from other regions of China) were based on China’s provinces. The remaining sequences out of the total have consisted of the cases from the other countries (13 Korea, 7 Japan, 14 USA, 8 Australia, 1 Belgium, 1 Finland, 6 France, 1 Germany, 2 Italy, 1 Nepal, 11 Singapore, 2 Thailand, 3 Taiwan and 2 UK). We also used the bat sequence of Yunnan as an outgroup to perform phylogenetic analysis in this study.
Multiple alignments and mutation detection: We used the sequence (NC_045512.2) registered in the GenBank, which was obtained from Wuhan Seafood Market as an outgroup to conduct this study to discover the origin and transmission history of SARS-CoV-2.
In total, 29903 base pair sequences were applied to align the other 126 sequences to the reference. The multiple alignments are performed by the BioEdit with a visual inspection [10]. To comprehend the similarity of the pair-wide genome sequence, we excluded the 5’ and 3’ 300 base-pair as it has a low sequencing quality. For the phylogenetic analysis, we also excluded the Wuhan 21 and the China 25 sequence since the mutation analysis of multiple alignments showed the China 25 sequence possessed 25 mutation sites, which could be regarded as a significant amount of mutation sites (2.8 times more than the average number of mutation sites).
Phylogenetic tree construction: The three methods (Maximum parsimony, Neighbor-Joining, Maximum likelihood) were used for this study. MEGA-X software is also used to compute the trees with 1000 bootstraps [11].
Results and Discussion
Mutation analysis based on multiple alignments and drawing the phylogenetic tree: The genome sequence of SARS-CoV-2 showed a 29870 base pair length without poly-A tails. The pair-wise similarity is 96.0% between human and bat sequences. Compared with the human genomes except for the Wuhan 21 genome (95.5%), which has low quality in the overall genome, its similarity is 99.9%–100%. In total, 177 mutation sites were detected through an analysis of 126 sequences (Table 2). Most of them were singleton mutations that occurred in a single individual’s virus genome. We observed that the only 30 mutations were shared with more than two other individual’s virus genome (Supplementary Table 1). As there are many similarity points, it may be inefficient to discriminate against the genome groups from the trees (Figure 1).
Genomic diversity of the virus genome: The first type of the virus genome is the No mutation group among the 30 mutation sites detected from 42 individual’s (17 of Wuhan, 10 China, 1 Korea, 3 Japan, 2 USA, 1 Australia, 1 Finland, 2 France, 1 Germany, 1 Singapore, 2 Thailand and 1 Taiwan). The second mutation type contained mutations at the two positions (8782, 28144) detected in 39 individual’s (Supplementary Table 2). The third mutation type contained one mutation at 26144 detected in 18 individual’s (Supplementary Table 3).
The mutations detected at the 15 positions are identified as being specific in 7 countries. Notably, we identified there are three specific mutations only represented in Korean samples: mutations at the position 4402 and 5062 in 8 Korean samples (Korea 02, 04, 05, 06, 07, 09, 10, 12), mutations at the position 26640 and 26677 in two Korean samples (Korea 03, 10), mutation at the position 2662 in three Japan samples (Japan 01, 02, 03), mutations at the position 614 and 5084 in two USA samples (USA 04, 05), mutations at the position 18488 and 23605 in two UK samples (UK 01, 02), mutation at the position 3177 in two USA samples (USA 04, 05), mutation at the position 22661 in three France samples (France 01, 02, 06), mutation at the position 27147 in two Singapore samples (Singapore 02, 06), mutation at the position 10138 in three Singapore samples (Singapore 05, 09, 10), and mutations at the position 28878 and 29742 in four Australia samples (Australia 03, 04, 05, 06). The remaining 12 mutations are classified as Chinese and belonged to other small countries.
As the sequence similarity is almost the same as 100%, it may bring up problematic implications for visualizing the cluster trees using a method associated with the Maximum parsimony tree [12]. We determined to use the Maximum parsimony tree for the phylogenetic analysis and implemented the only parsimonious informatics sites. Our result from the analysis showed that the Maximum parsimony tree could be employed to discriminate against the viral genome clusters. (Figure 1) showed a phylogenetic tree of 126 virus genomes using a Maximum parsimony tree. Generally, the clusters are divided into three clusters out of consideration for the tree. The group contained the most significant number of genomes is the no mutation group, which indicates the virus genome sequence is the same as the reference virus genome obtained from NCBI. The next largest group contained two mutations at the position 8782 and 28144. Interestingly, the sequences are more closely clustered to the bat virus genome obtained from China’s Yunnan region. The third group contained one mutation (26144), and it is more likely to be clustered close to the no mutation group.
Based on the mutation pattern that indicates the number of shared multi individual’s variants in each country, we detected that Korea and China’s cases had specific mutations on the 8782 and 28144 position. We notably identified mutations at the specific position (4402, 5062, 26640, 26677), which is transmitted by Wuhan’s city in China. We determined to disregard the other patterns as it might interrupt to predict the history of the virus transmission.
This study cannot cover the virus sequence from a small population size for Korean samples relatively. However, our study suggested that the analysis of deep sequence mutation would benefit the approach of well-understanding the virus transmission dynamic.
In terms of the COVID-19 diagnostic kit, the current diagnostic kit has simply detected whether the SARS-CoV-2 virus genome exists in the host sample [13]. We suggest that our findings provide a new diagnostic kit, including the detection and origin of the virus genome, by genotyping specific mutations from the population. In accumulating more samples to perform the analysis, it might have the possibility that the positions identified are no longer specific [14]. As for now, with current data samples, we were able to track transmission routes based on specific positions [15].
In conclusion, although our study did not use the novel sequence, our result provides evidence to deeply understand the virus genome mutation pattern. We hope our study and its applied method will be of critical importance and robust references used for the further COVID-19 research and prevention of the transmission speed. The follow-up research with this study’s information will be imperative for researchers to dive into vaccine development and disease control.
Keywords
Severe acute respiratory syndrome; Coronavirus (SARS-CoV); Wuhan coronavirus; Phylogeny; Whole genome sequence; Human-to-human transmission
Cite this article
Kim H, Jeong S, Wang Y, Kim JO, Kim T, Hong KW. Mutation pattern analysis of the public SARS-CoV-2 genome sequences. Clin Case Rep J. 2020;1(6):1–5.
Copyright
© 2020 Kyung-Won Hong. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
