Abstract
Hereditary Hypophosphatemic Rickets with Hypercalciuria (HHRH) is a rare autosomal recessive form of hypophosphatemic rickets. It is caused by mutations in the SLC34A3 gene, which encodes the renal sodium-dependent phosphate cotransporter 2c (NaPi-IIc). It is characterized by normal plasma levels of calcium and 25-hydroxyvitamin D, an elevated level of 1,25-Dihydroxyvitamin D [1,25(OH)2D], a low level of parathyroid hormone, hypophosphatemia, hyperphosphaturia, hypercalciuria, and a low level of plasma fibroblast growth factor 23 (FGF23). The presence of hypercalciuria, raised plasma 1,25(OH)2D, low plasma FGF23, and nephrocalcinosis helps differentiate HHRH from other hypophosphatemic forms of rickets. We report a rare case of HHRH in a 9-year-old girl who presented to our institute to evaluate short stature. The patient was found to have craniosynostosis with no clinical or radiological signs of rickets. She had classical biochemical features of HHRH, and a molecular genetic study revealed a large homozygous deletion encompassing part of exon 4, all of intron 4, all of exon 5, and part of intron 5 of the SLC34A3 gene. This created a premature stop, c.284_449-23del (p. ser95), and confirmed the diagnosis of HHRH. This case highlights the importance of investigating a patient who presents with hypophosphatemia even with no clinical, or radiological features of rickets.
Introduction
Hereditary Hypophosphatemic Rickets with Hypercalciuria (HHRH) is a rare autosomal recessive genetic disorder (OMIM #241530) caused by a mutation of the SLC34A3 gene. This gene encodes the type 2c sodium phosphate co-transporter (NPT2c), responsible for the reabsorption of filtered phosphate in the proximal renal tubules [1]. The result is excessive renal loss of phosphate, and phosphaturia leads to low plasma levels of fibroblast growth factor 23 (FGF23).
The resulting hypophosphatemia led to the higher renal 1-alpha hydroxylase enzyme activity and increased renal synthesis of 1,25-Dihydroxyvitamin D [1,25(OH)2D]. The increased serum level of 1,25(OH)2D results in hyperabsorption of calcium from the gastrointestinal tract and suppresses parathyroid hormone production (PTH), leading to hypercalciuria and an increased propensity toward nephrocalcinosis or nephrolithiasis. Thus, classical biochemical findings in patients with HHRH include normal plasma levels of calcium and 25-hydroxyvitamin D, an elevated level of 1,25-Dihydroxyvitamin D, a low level of parathyroid hormone, hypophosphatemia, hyperphosphaturia, and hypercalciuria [1].
It is difficult to differentiate HHRH from other types of hereditary hypophosphatemic rickets. However, the presence of hypercalciuria, nephrocalcinosis, and low plasma levels of FGF23 usually help to differentiate it from other forms of hypophosphatemic rickets, such as X-linked hypophosphatemic rickets, autosomal dominant hypophosphatemic rickets, and autosomal recessive hypophosphatemic rickets, which are treated with phosphate and calcitriol supplementation. The hypercalciuric state of patients with HHRH may worsen if they are treated with calcitriol.
This study describes a unique case of a 9-year-old Omani girl who presented with short stature and craniosynostosis. She had no clinical or radiological features of rickets. Biochemical and genetic investigations support a diagnosis of HHRH.
Case Report
A 9-year-old Omani girl presented for specialized evaluation to the Pediatric Endocrinology Department at the National Diabetes and Endocrine Center (NDEC), Royal Hospital, Oman. She was 5-year-old at the time. She presented with her parents for a short stature complaint, which they noted because she was shorter than her youngest sister and her peers at school.
Past medical history: The patient’s parents were non-consanguineous. She was born by spontaneous vaginal delivery at full-term with good Apgar scores. Her birth weight was 2.4 kg (in the 3rd percentile on the standard WHO growth chart), her birth length was 48 cm (50th percentile on the WHO growth chart), and her head circumference was 32 cm (10th percentile on the WHO growth chart). She had an uneventful prenatal, natal, and postnatal history, and her development had proceeded normally.
The patient had no history of chronic illness and was not using any medications. She was noted to have an abnormal head shape at the age of one year, for which she was evaluated at another hospital, and her parents were reassured that the shape was normal. The child had no history of fracture or bony pain, and clinically, there were no bony deformities.
Her nutrition was normal, and her milk intake was one cup of fresh milk (around 200 ml) per day (Al-Marai milk contains only 40 IU of vitamin D3 per 100 ml). She has four sisters, and all of them are healthy, but the eldest one (11-year-old) was also short (below the third percentile). The family had a short stature history (her paternal grandfather, both parents, and her oldest sister). There was no family history of hematuria, renal stones, rickets, or bone deformities.
Clinical examination: The patient looked well, was active, and had no clear dysmorphic features. On anthropometrical evaluation, her height was 87.9 cm (below the 3rd percentile with –4.4 SD on the WHO growth chart), her weight was 12 kg (below the 3rd percentile with –3SD on the WHO growth chart), and her head circumference was 46 cm (below the 3rd percentile with –3SD on the WHO growth chart). Her mid parental height (MPH) was 153 cm (below the 3rd percentile on the WHO growth chart), her upper segment to lower segment (US/LS) ratio was 0.9, and her growth rate for one year was 4.5 cm.
Her blood pressure was 105/55 mmHg, which is normal for her age, sex, and height. She was noted to have scaphocephaly. She had normal teeth with no clinical features of rickets or deformities. The ophthalmic examination was normal, with no signs of papilledema.
Laboratory investigations: The patient’s full blood count showed normal hemoglobin with other parameters normal, as well as normal renal function, normal liver function, normal thyroid function, and negative celiac screening. Her serum IGF-1 level was 4.8 nmol/L (NR: 3.5–36.7). A dynamic growth hormone (GH) stimulation test (clonidine and glucagon) showed a good response with peak serum GH levels of 25.19 mIU/L for clonidine and 16 mIU/L for glucagon (normal range (NR) response: more than 20 mIU/L).
Her serum concentration of adjusted total calcium was at the upper end of the reference range (2.67 mmol/L; NR: 1.9–2.70), her serum level of phosphate was low for her age (0.91 mmol/L; NR: 1.0–1.95), and her serum alkaline phosphatase (ALP) was raised for her age (554 IU/L; NR: 90–201). She had a normal 25-hydroxyvitamin D level of 63.6 nmol/L (NR: 50–150) and a low PTH level of 1.10 pmol/L (NR: 1.6–6.9). Her serum level of (1, 25(OH)2D) was 226 pmol/L (NR: 43–168), and her random urinary calcium-to-creatinine ratio was 1.79 (NR: 0.04–0.8).
The patient had low tubular phosphate reabsorption (TRP) of 68% and a low maximal TRP/glomerular filtration rate (TmP/GFR) of 0.65 (NR: 1.15–2.4). Her level of intact/c-terminal plasma FGF23 (Cerba Laboratoire, France) level was < 21 ng/L (NR: 25–50). The results of her initial and subsequent biochemical tests are presented in (Table 1).
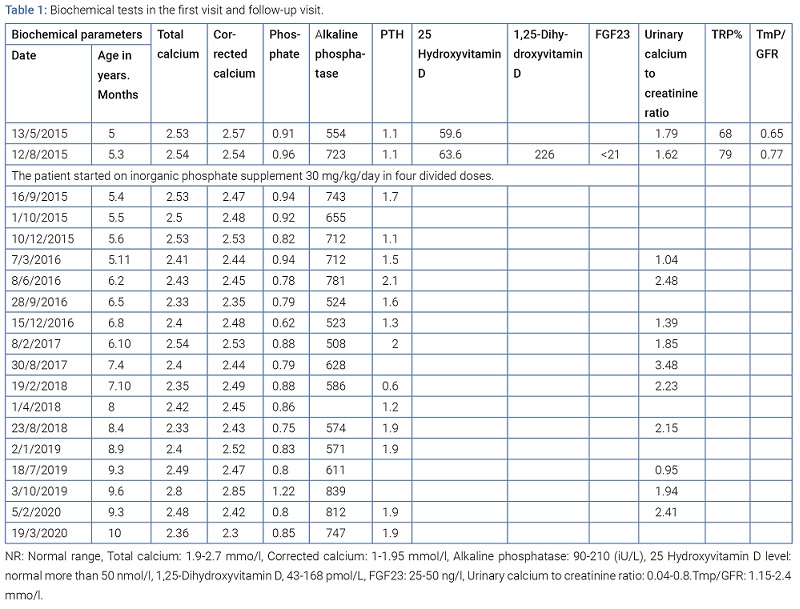
Radiological examination: Radiographs of the wrist and knees showed mild osteopenia with no rickets’ radiological features (Figure 1).
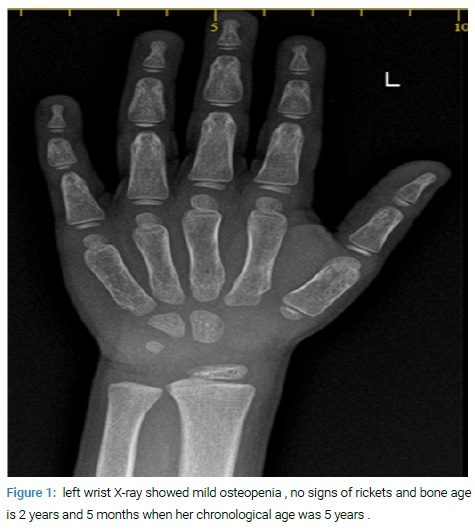
The Computed Tomography (CT) scan of her head showed premature closure of the sagittal suture resulting in scaphocephaly, but her brain was normal (Figure 2).
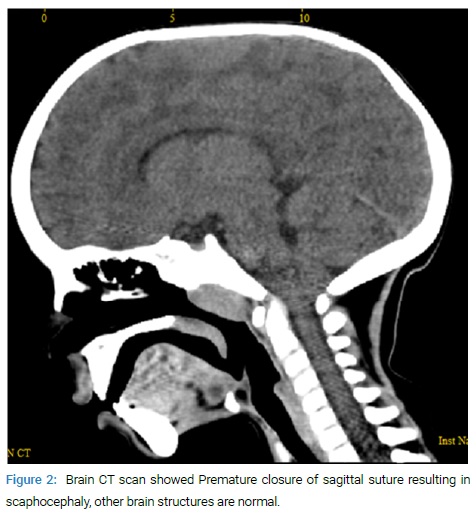
The kidneys’ ultrasonography showed bilateral diffuse medullary nephrocalcinosis, right kidney length of 7.2 cm, left kidney length of 7.5 cm, and no calculi or hydronephrosis (Figure 3).
Hypophosphatemic rickets with hypercalciuria was suggested by the patient’s biochemical findings (hypophosphatemia, normocalcemia, high ALP, suppressed PTH, high [1,25(OH)2D], low FGF23, hypercalciuria with low TRP, low Tmp/GFR) and radiological features of nephrocalcinosis. Whole-exome sequencing of the patient’s DNA was performed by the Centogene Laboratory (Germany), which revealed a large homozygous deletion encompassing part of exon 4, all of intron 4, all of exon 5, and part of intron 5 of the SLC34A3 gene. This created a premature stop, c.284_449-23del (p. ser95), and confirmed the diagnosis of HHRH.
Treatment: The child was started on oral inorganic phosphate supplementation (Sandoz) at a dose of 30 mg/kg/day, which was administered in four divided doses. She did not develop side effects of inorganic phosphate therapy (abdominal pain, diarrhea, or abdominal distension). In view of her hypercalciuria and nephrocalcinosis, she was started on oral hydrochlorothiazide at 12.5 mg once daily. Due to the poor taste of the inorganic phosphate therapy, the adherence to the treatment was poor. Her hypercalciuria persisted, and she was not taking vitamin D or calcium supplements. The patient drank one cup of fresh milk (around 200 ml) (Al-Marai milk, a product of Saudi Arabia), in which each 100 ml contains only 40 UI of vitamin D3.
The parents received full counseling about the disease, the prognosis, and the need to adhere to oral phosphate therapy. The parents and their four sisters were investigated biochemically for bone profiles, parathyroid hormone, urinary calcium creatinine ratio, and renal ultrasound. All results were within the reference ranges apart from the older sister (age 11 years), who was found to have biochemical features favoring HHRH but no nephrocalcinosis and no radiological features of rickets apart from osteopenia. Her lumbar spine (L1–L4) areal bone mineral density (BMD) z-score was measured by dual X-ray absorptiometry (DXA), and the result was normal for her age.
Discussion
We have described a case of HHRH in a 9-year-old girl who presented with short stature and craniosynostosis. She was found to have hypophosphatemia due to renal phosphate wastage, corrected total calcium at the upper end of the reference range, high alkaline phosphatase, suppressed PTH, normal 25-hydroxyvitamin D, high [1,25(OH)2D], low FGF23 levels, hypercalciuria, and nephrocalcinosis. However, she did not have clinical or radiological features of rickets. Genetic analysis indicated a diagnosis of HHRH. This study is the first case of HHRH to be reported in the Omani population to the best of our knowledge.
HHRH is an autosomal recessive disease and is caused by a loss-of-function mutation in the SLC34A3 gene. This gene consists of 13 exons with an initiation codon located in exon 2 (R = 5). SLC34A3 encodes sodium phosphate co-transport protein IIc (NaPi-IIc or NPT2c), a multi-pass transmembrane protein that actively transports phosphate into cells by Na co-transport in the renal brush border membrane [1]. Individuals with HHRH carry compound heterozygous or homozygous loss-of-function mutations in the sodium-phosphate co-transporter NPTC2c.
The mutation results in the development of urinary phosphate (Pi) wasting, hypophosphatemic rickets, bowing, short stature, and appropriately elevated 1,25(OH)2D levels. These features set this FGF23-independent disorder apart from the more common X-linked hypophosphatemia. The elevated 1,25(OH)2D levels result in hypercalciuria due to enhanced intestinal calcium absorption and reduced PTH-dependent calcium-reabsorption in the distal renal tubules, leading to the development of kidney stones/nephrocalcinosis [2].
In most other forms of congenital hypophosphatemia, there are high levels of FGF23, which cause both renal phosphate wasting and inhibition of 1-alpha hydroxylase activity, resulting in low1,25(OH)2D. However, HHRH is not related to an abnormality of FGF23 action, and typically, levels are normal or low. Consequently, 1,25(OH)2D production is not inhibited, such that hypercalciuria may result [2].
Many reported cases with homozygous mutation and compound heterozygous mutations in SLC34A3 describe the phenotype of 56 patients from 34 families [1–3]. The first cases were described in 1985 in a large consanguineous Bedouin Arab Kindred, in which Tidernad et al. recognized that this disorder is different from the common X-linked hypophasemic rickets (XLH, OMIM 30780) [1,2]. The prevalence of HHRH has been reported to be 1:250,000, and it is less frequent than XLH (1:20,000) [2].
Our reported case exhibited biochemical features of HHRH but no skeletal abnormalities of rickets apart from craniosynostosis. However, the genetic analysis confirmed the diagnosis and showed a homozygous mutation of SLC34A3. HHRH without radiological evidence of rickets has previously been described with homozygous and compound heterozygous mutations of the SLC34A3 gene [4,5].
Our patient’s siblings and parents were investigated biochemically and radiologically. Only one sibling, an 11-year-old girl, was found to have biochemical features suggestive of HHRH, but there was no evidence of rickets or osteomalacia apart from short stature and no nephrocalcinosis. Her DXA-measured L1-L4 BMD Z-score was normal for her age. The parents and siblings showed normal biochemical findings with no nephrocalcinosis evidence in the abdomen ultrasound apart from the sibling mentioned. Mutational analysis is required to determine the other sibling’s heterozygous carrier status, but this was not done because of the limitations of our lab. Patients with HHRH can present with short stature, as in our patient. This could be attributed partially to her disease and to her strong family history of short stature as both parents and her paternal grandfather are short. According to the WHO chart, her mid parental height is below the 3rd percentile, and she is still prepubertal.
Our patient was found to have craniosynostosis with no other structural anomalies of the brain. She is developmentally normal and has excellent academic performance in school. She was evaluated by an ophthalmologist, who reported that she has no papilledema.
The patient started only inorganic phosphate treatment, but her adherence to the treatment was poor even though she did not develop gastrointestinal side effects from this medication. Hence, hypercalciuria occurred, which was not related to excessive dietary intake of vitamin D or calcium-rich dairy products.
This case highlights the variability in the clinical presentation and biochemical profile of HHRH. It also shows the importance of correct diagnosis because orally administered inorganic phosphate is usually adequate to treat the disorder, in contrast to other types of hypophosphatemic rickets, which require vitamin D supplements. This case also illustrates that it can be challenging for children to adhere to inorganic phosphate therapy, which can reduce hypercalciuria and thus prevent the progression of nephrocalcinosis.
Thus, there is a need for more palatable inorganic phosphate preparations. Burosumab, a humanized monoclonal antibody to FGF-23, has been licensed for the treatment of XLH but is not appropriate for HHRH as it is not an FGF23-dependent cause of chronic hypophosphatemia [6]. The real prevalence of HHRH needs to be studied further with consideration of the importance of proper diagnosis and genetic counseling for families with this disorder.
Cite this article
Al-Badi MK, Al-Senani AM, Mughal MZ. A case of short stature and sagittal craniosynostosis in an Omani girl with hereditary hypophosphatemic rickets with hypercalciuria. Clin Case Rep J. 2021;2(1):1–5.
