Abstract
Orbital granulocytic sarcoma involving the central nervous system and coinciding with acute megakaryoblastic leukemia is a rare complication of this disease. We report a case of a previously healthy male infant 20-month-old who developed acute megakaryoblastic leukemia and in whom the initial diagnosis was difficult because of associated severe myelofibrosis. We focus on this report on the clinical and cyto-histological features and problems concerning differential diagnosis and the importance of risk group stratification to identify specific relapse risks and select the most appropriate treatment, intensive-timing chemotherapy regimen, or HSCT obtain good outcomes.
Introduction
Granulocytic Sarcoma (GS), previously known as chloroma, is a solid tumor of myeloid precursor cells described for the first time by Burns in the early 19th century and originally termed chloroma by Knight in 1853 due to its green color attributed to the enzyme Myeloperoxidase (MPO). Later changed to GS by Rappaport in 1966, following reports of cases in which the tumor was not green and had sarcoma’s gross features [1–3]. This led to frequent misdiagnosis as high as 75% in a historical study in 1986 and lower incidence (25%–47%) in more recent reports. However, the diagnosis of GS in the absence of myeloid leukemia still poses a diagnostic challenge, and most patients were mistaken as suffering from malignant lymphoma [4–11]. Since Dock first noted the association of GS with Acute Myeloid Leukemia (AML) the tumor has been reported more frequently in patients with AML, especially in children and is described as a solid tumor of immature granulocytic precursor cells, not always composed of granulocytes so terms as extramedullary tumors or extramedullary leukemia have also been proposed [12–17]. It presents as an extramedullary mass, and a manifestation of any myeloproliferative disorder that most frequently affects the bones of the skull, sternum or ribs, peritoneum, lymph nodes, and occasionally soft tissues around the dura, sinuses and orbit, and is included as one of the major subgroups of myeloid neoplasms and acute leukemia in the WHO classification [3,11,14,18] GS is more common in pediatric patients, especially younger than 15 years old, and many cases have been reported in the head and neck region, but the most frequent presentation is unilateral exophthalmos due to orbit involvement (OGS) and occurs associated with Acute Myeloid Leukemia (AML), mainly AMLM2 or AML with a monocytic component (M4 or M5) [10,17,19–27].
Case Presentation
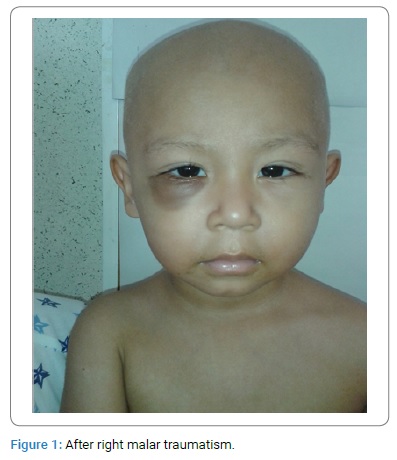
A previously healthy 20-month-old male presented with a 3-day history of fever and upper tract infection, for which antibiotic treatment was given, and fever and malaise subsided. In the following weeks, the patient developed progressive pallor and respiratory symptoms. He was seen two months later by his treating physician; physical examination revealed tachypnea, a grade III/IV systolic ejection heart murmur, and hepatomegaly 2 cm below the right costal margin. Complete blood count (CBC) reported hemoglobin (Hb) 5.2 gm/dL, hematocrit 17%, white blood cell count (WBC) 7.3 x 109/L and differential leukocyte count showed 1.3 x 109/L absolute neutrophils, 77% lymphocytes, 4.2% monocytes and 76 x 109/L platelets. He was transfused with packed red blood cells 20 ml x kg on two occasions to correct anemia and respiratory symptoms and was referred to our hospital for further evaluation and admitted to the outpatient hematology department four weeks later, where a diagnostic work-up was established. At that time, the patient was asymptomatic and physical examination unremarkable; a CBC reported Hb 16.6 gm/dL, hematocrit 45.7%, WBC 6.5 x 109/L, and differential leukocyte count showed 2.1 x 109/L absolute neutrophils, 55% lymphocytes, 10% monocytes, and 46 x 109/L platelets. Blood chemistry, coagulation, iron and virus profiles (CMV, HV, EBV, Parvovirus), urinalysis and culture, folic acid, and vitamin B12 were normal or negative. A Bone Marrow Aspiration (BMA) showed scarce cellularity, absent megakaryocytes, lymphocyte predominance, myeloid maturation arrest with moderate granular asynchronism, and negative for hematologic malignancy. A repeated BMA yielded a dry tap, and a bone marrow biopsy yielded scarce material; extended screening tests looking for an underlying immune disease process was carried on, and he continued follow-up in the ambulatory hematology clinic. One month later, he was admitted to the emergency ward after a malar traumatism and facial swelling; ophthalmic examination revealed bilateral upper and lower eyelid oedema, probably secondary to bone traumatism. Ocular movements were normal; no signs of periorbital cellulitis and oedema disappeared after 24 hours of hospitalization (Figure 1).
Plain RX of facial bones was normal. Physical examination revealed hepatomegaly 2-3-2 cm and splenomegaly 2 cm below the costal margin, and the test was negative. A CBC showed Hb 9.8 gm d/L, hematocrit 29% WBC 11.2 x 109/L, 5.4 x 109/L absolute neutrophils, and differential leukocyte count 36% lymphocytes, 14% monocytes, and 14 x 109/L platelets, and isolated blasts with cytoplasmic protrusion or blebs and 1.4% reticulocytes. CBC showed progressive anemia (Hb 9.8 gm/dL), thrombocytopenia (14 x 109/L), a small number of circulating blasts, and splenomegaly. BMA was performed, which yielded a dry tap, and bone marrow biopsy reported myelofibrosis (Figure 2, Figure 3); all other laboratory tests, including BUN and uric acid, were reported normal; he was transfused with packed red blood cells and discharged to continue follow-up in the outpatient hematology department.
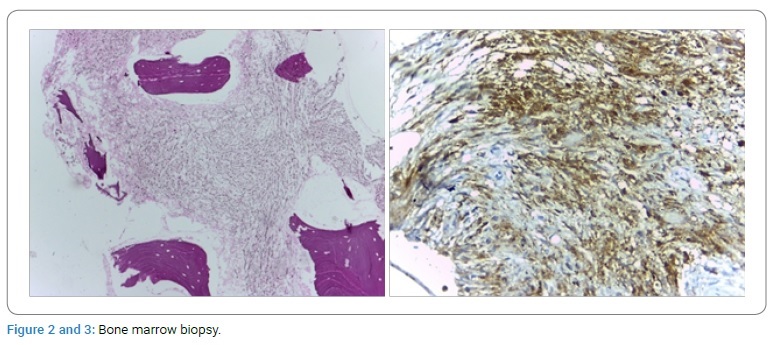
Histologic features: Decreased cellularity due to diffuse infiltration of the marrow by fibrous connective tissue replacing bone marrow elements. Special histochemical staining with tri-chrome Masson revealed collagen deposits with few scattered erythroid and myeloid precursors and dysplastic megakaryocytes.
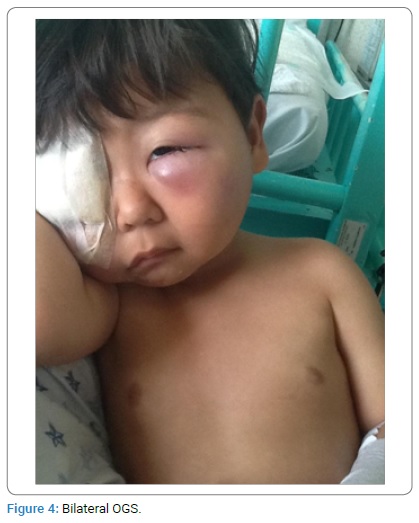
Two weeks later, he was seen in the outpatient department to present a rapidly progressive bilateral ocular proptosis, right eye pain, epiphora, chemosis, and severe bilateral upper and lower eyelid oedema (Figure 4), a left cervical enlarged lymph node, and pancytopenia. Blood smear analysis showed a steady increase in the number of circulating blasts during the following week (5%–10%–30%), some with cytoplasmic protrusion or blebs; BMA and the immunologic profile established the diagnosis of AMKL by the expression on blast cells of platelet specific markers CD41 (78%), CD42 (68%) and CD61 (98%) by multiparameter flow cytometry. Bone marrow biopsy was reported compatible with acute megakaryoblastic leukemia. Six hours after BMA and biopsy, the patient was admitted to the hematology ward because of continuous bleeding in the puncture site, which required treatment with platelet concentrates, and an urgent diagnostic work-up was programmed that included cranial simple and contrasted computed tomography (CT) scans and magnetic resonance imaging (MRI) of both orbits, excisional biopsy of the retroorbital tumor and left cervical excisional biopsy.
Excisional OGS revealed GS with megakaryoblastic differentiation (Figure 5), and the left cervical excisional lymph node revealed a granulocytic sarcoma (Figure 6).
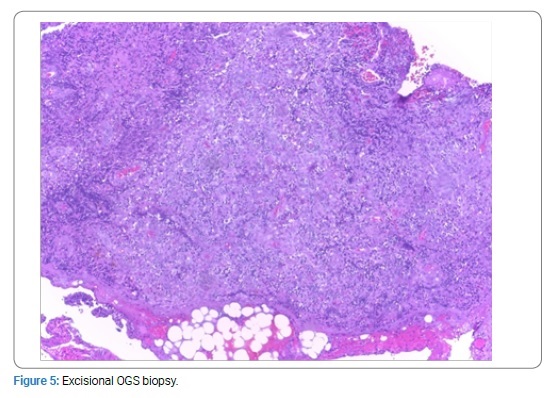
Fragment of soft tissues with a neoplastic infiltration of medium to large blast cells with a variable nucleocytoplasmic ratio, an oval nucleus with fine stippled chromatin and 1 to 3 inconspicuous nucleoli forming a sheet cell lamina and numerous mitosis.
Microscopic description: GS with megakaryoblastic differentiation. Immunohistochemical staining was positive for CD45 (+++ ) and VIM (+++) diffuse, CD34 and CD31 positive focal and irregular, LMP-1 positive. Negative markers. CD3, CD20, CD30, CD15, CD1a, PS 100, CD21, CKSAE1/AE•, CD163, CD68, ALK, MPO, CD117, CD99. LMP-1 marker is frequently positive in bone marrow megakaryocytes of patients without a previous history of EBV infection and this expression is seen in undifferentiated megakaryocytes.
MRI revealed a large irregular tumor with bilateral infiltration of upper jaw bones and orbits and extending to left posterior fossa (Figure 7, Figure 8).
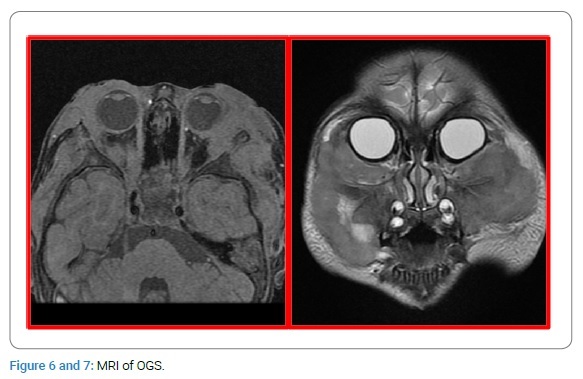
Axial T1-weighted and Coronal T2-weighted SPIN ECO revealed an irregular homogeneous huge tumor infiltrating both orbits, bilateral malar, temporal areas and maxillar bones, of left predominance and extending into the posterior cranial fossa.
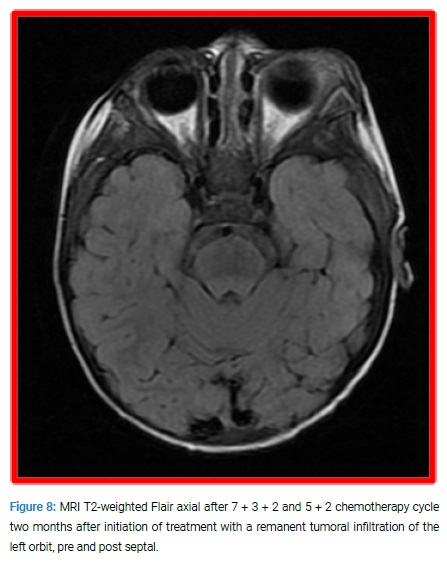
The patient began an induction AML protocol with a 7 + 3 + 2 (cytosine arabinoside, daunorubicin, etoposide), an induction regimen, and triple intrathecal therapy (methotrexate, Ara-c, hydrocortisone) twice a week for six doses and a 5 + 2 cytosine arabinoside and daunorubicin cycle. Hospitalization was complicated by anemia, bleeding, neutropenia, and systemic inflammatory response syndrome, which responded well to packed red cells, platelet concentrates transfusions and IV broad-spectrum antibiotics.
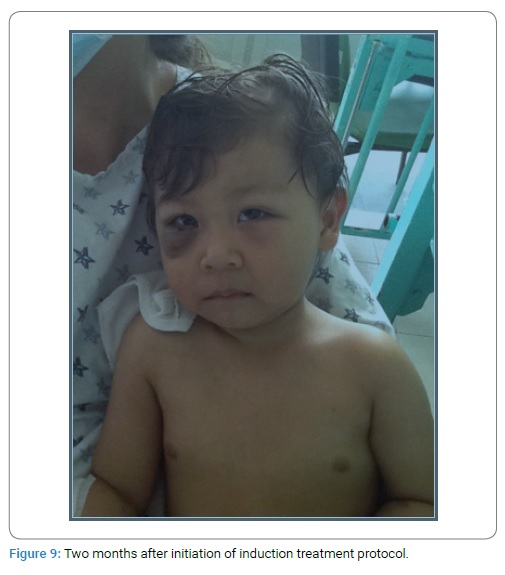
Two months after chemotherapy initiation, there was a significant clinical improvement with prompt resolution of both retroorbital and cerebral tumors, and the patient attained complete remission three months later (Figure 9).
He continued consolidation therapy with high dose Ara-c (HDAC) + TIT and L-asparaginase rescue (Capizzi type II) for two cycles and etoposide+ Ara-c two cycles with triple IT therapy. Maintenance therapy consisted of multiple combined chemotherapy with TAAP, COAP, POMP 5-day treatment and 9 days to 14 days rest and periodic triple IT therapy. After two years in Continuous Complete Remission (CCR), elective cessation of chemotherapy was programmed. He continues in CCR five years after stopping therapy.
Discussion
GS are rare solid tumors of immature myeloid precursor cells that presents as an extramedullary mass or a manifestation of myelodysplastic syndrome, chronic myeloid leukemia or any myeloproliferative disorder, and has been reported with an incidence of 2/1.000.000 in adults or 3%–9.1% in myeloproliferative disorders [4,11,28–30]. In the retrospective study of Dusenbery et al. of the Children´s Cancer Group of 1832 patients with newly diagnosed AML, 199 (10.9%) had evidence of extramedullary leukemia, and these 12 (0.65%) corresponded to orbital GS. In contrast, Johnston et al. in a more recent study of the Children Oncology Group, reported that out of 1459 analyzed pediatric patients with AML, 99 (7.3%) presented myeloid sarcoma., 1% CNS sarcoma, and 2% OGS [17,31]. The mean age of presentation in childhood varies between 7 years and 8.8 years [23,32,33]. Orbit GS (OGS) has been reported to be more prevalent in countries from Asia, Africa, and Latin America, [32–36] and occurs associated with acute myeloid leukemia (AML), mainly AML-M2 with t (8;21) (q22;q22) or AML with a monocytic component (M4 or M5) [10,17,19–25,37].
GS can precede acute leukemia (de novo), present concurrently or during the disease (relapse) [3,5,9,10,12,23]. When GS occurs in patients with known AML or other myeloproliferative disorder, there is usually a little difficult for the clinician to establish and for the pathologist to make a correct diagnosis, but when GS precedes the development of AML or when it develops in a subtype of AML-AMKL-that frequently presents with severe myelofibrosis, diagnosis can be extremely difficult [3,5,8,9,11]. In our case, although the onset of symptoms, initial laboratory features, and a high clinical awareness suggested acute leukemia, it took four months to establish a definitive diagnosis. We attributed this to the slowly progressive clinical course of the disease extending over months, the presence of severe myelofibrosis, repeated BMA resulting in dry taps, and a late clinical event resembling “blast crisis” of chronic myeloproliferative disorders, as indicated by a steady increase in the number of blast cells in peripheral blood and BMA smears observed in the last month of follow-up, similar to the evolution of some cases of childhood AMKL as previously reported [38–40].
OGS is a rare event, and its presence in patients with a previous myeloproliferative disorder suggests an impending blast crisis or leukemic transformation, or the initial manifestation of relapse in a previously treated AML patient in remission, but in patients, without known hematologic diseases, it should alert clinicians to start a thorough, immediate search of AML, non-Hodgkin or histiocytic lymphoma, metastatic neuroblastoma, rhabdomyosarcoma, and small round blue cell tumors, by BMA and biopsy, tissue biopsy stained with H&E, and immunohistochemical methods, multiparameter flow cytometry, FISH, CT, and MRI scans, FDG-PET CT for an early diagnosis and prompt and optimal treatment.
One of the first cases of OGS and intracranial tumors in AMKL was reported by Slavic et al. in 1991 in an 8-month child who responded well with a sharp reduction of intracranial masses two weeks after initiation of antileukemic therapy. However, unfortunately, the patient died of bacteremia 5 weeks after starting treatment. More recently, the case of Baldwin and Mian, that had a complete response to chemotherapy and local radiotherapy [24,41]. Although OGS is a rare event, in a series of 633 patients with AML treated in SJCRH over 30 years, 30 (4.7%) had one or more at some point in the course of their disease, and 13 (2%) had OGS, but despite its rarity, subcutaneous tissue and orbit were the most frequently affected sites. Another interesting issue of the report was that the tumor resolved completely within three months in 14/31(45.2%) patients treated with chemotherapy alone [42], as in the present case. Leukemic involvement of the central nervous system was common before effective chemotherapy protocols and was the first site of relapse in nearly 50% of children in remission previous to the incorporation of prophylactic therapy to the CNS in posterior protocols [43]. Masses within the brain are uncommon and were reported occasionally in acute leukemia until the middle of 1990s when two reviews and a casuistic of patients referred to its higher frequency in AML, its radiologic features, and highlighted the importance of early chemotherapy and radiotherapy to treat these complications [29,31,43–46].
It is difficult to speculate why our patient responded so well after the initiation of chemotherapy. May be because he was treated aggressively, or because his leukemia was not inherently aggressive, or because of both factors is not clear. Since the early 1990s, and based on different response rates observed in early-onset AMKL in infants with Down´s syndrome and t (1;22) (p13; q13) or later in life, investigators were intrigued and started wondering if this was one or several diseases [47]. Although several subsequent reports indicated that the t (1;22) (p13;q13) was a nonrandom marker and restricted to infant AMKL, but that some cases expressed clinical and genetic heterogeneity, similar to some patients in our country [48–51]. In 2002 the Groupe Français de Cytogénétique Hématologique identified nine cytogenetic groups in childhood and adult megakaryoblastic leukemia, confirming different subtypes of the same disease, four of which were exclusively composed of children: (group 1) patients with Down’s syndrome, (group 2) numerical abnormalities only, (group 3) t (1;22) (p13; q 13) (group 4) t (9;22) (q34; q11).
In the last decade, several groups of investigators have identified aggressive subtypes of pediatric AMKL, and four years ago, de Rooij et al. pointed out that recurrent abnormalities identified in nearly half of the patients by deep sequencing studies could be used for risk group stratification and included as a low/intermediate risk factor t (1;22) (p13; q13) resulting in a chimeric fusion RBM15/MKL1 and patients without any of the four specified cytogenetic abnormalities and patients with the CBFA2T3/GLIS2, rearranged KMT2A, NUP98/KDM5A, and monosomy 7 as high-risk factors in AMKL [52–55].
In conclusion, we can only speculate if the good response in our patient was the result of early intensive treatment as has been reported recently by the Children´s Oncology Group in patients with AML treated with an intensive-timing chemotherapy regimen which showed an excellent outcome in OGS and CNS GS or because he had an inherently less aggressive AMKL, or both factors [31].
Cite this article
Paredes-Aguilera R, Santiago NL, Méndez-Meraz A, López-Corella E, De Uña-Flores A, Lina Romero-Guzmán L. Orbital granulocytic sarcoma presenting simultaneously with acute megakaryoblastic leukemia. Clin Case Rep J. 2021;2(2):1–7.
