Abstract
The etiology of perinatal death often remains unidentified, yet proper diagnosis is crucial for appropriate family counseling and future pregnancy planning. We present a neonate delivered at 30 weeks and four days gestational age in the setting of hydrops fetalis and intrauterine growth restriction to non-consanguineous parents. Gross morphology was consistent with Fetal Akinesia Deformation Sequence (FADS) in addition to the hydropic changes. The infant expired at 9 minutes of life. Evaluation with whole-exome sequencing identified compound heterozygosity for TTN gene mutations: a paternally inherited substitution (c.36253C > T p.Q12085X) and a maternally inherited deletion (c.38919delC p.L12974WfsX104). In addition, mosaic monosomy X was identified, providing a likely etiology for the fetal hydrops. The trio exome sequence demonstrated predicted loss of function mutations in the fetal isoforms of TTN from both parents, raising the possibility that this gene played a role in the fetal phenotype. A literature search identified a series of other cases in which recessive TTN mutations resulted in a similar arthrogryposis phenotype, supporting the claim that TTN variants played an etiologic role in developing FADS in the current case. This case illustrates the value of whole-exome sequencing in the evaluation of fetuses and neonates with multiple malformations.
Abbreviations
FADS: Fetal Akinesia Deformation Sequence; TTN: Titin Gene (TTN).
Introduction
The workup for intrauterine fetal demise and neonatal death aims to identify contributing factors to explain the cause of death and address risk in future pregnancies. We describe the use of whole-exome sequencing in the evaluation of a neonate with a complex phenotype, including Fetal Akinesia Deformation Sequence (FADS), hydrops, and neonatal demise. The classic clinical presentation of FADS includes intrauterine growth restriction, joint contractures, pulmonary hypoplasia, abnormal facies, and camptodactyly. The same or similar constellation of findings has also been described as Pena-Shokier syndrome, arthrogryposis multiplex congenita, multiple pterygium syndrome, and lethal congenital contracture syndrome [1,2]. The causes of this group of related disorders are myriad and include genetic and environmental factors. This case provides evidence for the utility of whole-exome sequencing in identifying genetic causes of neonatal and perinatal demise and adds to the growing spectrum of phenotypes associated with TTN gene mutations.
Case Presentation
A 26-year-old G1P0 with no significant medical history was followed in our clinic for prenatal care. Ultrasound was performed to confirm gestational age, and routine laboratory tests were normal. In addition, an ultrasound at 20 weeks showed normal fetal anatomy.
The patient’s clinical course was unremarkable aside from concerns from the patient that she did not feel fetal movement, for which no additional testing or imaging was ordered, as the 20-week scan had been normal. At 28 weeks, the fundal height was noted to be 34 cm, suggesting macrosomia and/or polyhydramnios. Ultrasound at 30 weeks and four days (Figure 1) demonstrated a growth-restricted fetus (1st percentile) with severe hydropic changes, flexion contractures, and polyhydramnios (amniotic fluid index 24).
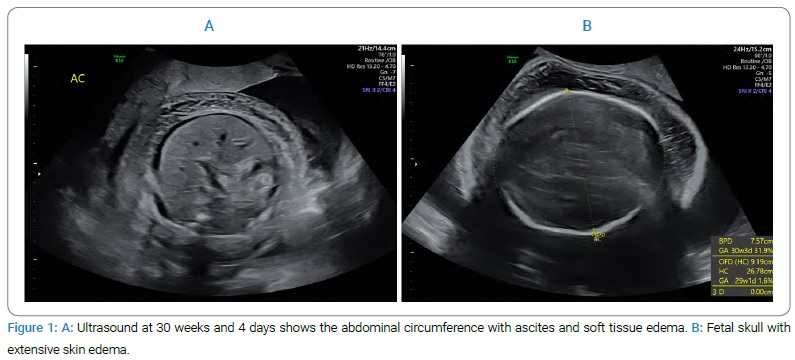
No fetal motion was seen. A category III fetal heart tracing was noted following the ultrasound examination, and an immediate cesarean delivery was performed. Gross inspection of the neonate revealed a severely edematous female infant with friable skin and multiple flexion contractures. APGAR scores, an indicator of neonatal well-being, were 1 and 1, at one minute and five minutes of life, respectively (out of maximum score possible score of 10). An unsuccessful attempt was made at resuscitation, and the infant expired at 9 minutes of life. The mother recovered uneventfully.
An autopsy revealed multiple joint contractures with pterygia of bilateral upper and lower extremities, severely underdeveloped skeletal muscle, bilateral humerus fractures, hypoplastic lungs with bilateral effusions, ascites, hypoplastic heart, triangular facies with retrognathia, short neck, absent palmar creases, and rocker-bottom feet. The findings were deemed consistent with FADS; however, the hydropic changes were considered atypical for this diagnosis.
The whole-exome sequence was performed on the mother, father, and fetus by GeneDx using the Illumina platform. Trio-WES revealed reduced X chromosome read counts, consistent with mosaic monosomy X, as well as two likely pathogenic, loss-of-function variants in the TTN gene. A paternally inherited substitution (c.36253C > T p.Q12085X) predicting a premature stop codon within exon 168 and a maternally inherited deletion (c.38919delC p.L12974WfsX104), which introduces a frameshift and subsequent stop codon within exon 200. Results were confirmed via Sanger sequencing. Although both variants were clearly predicted to disrupt protein function, neither was present in ClinVar or Genome Aggregation Database (gnomAD) and therefore, both were described as “variants of uncertain significance; likely pathogenic.”
Discussion
We report a neonate, born by emergency Cesarean section at 30 weeks gestation to non-consanguineous parents, that died immediately after birth with an unusual phenotype consisting of FADS as well as hydrops fetalis. An autopsy confirmed the clinical impression of fetal akinesia sequence and hydrops but did not provide an etiology for this constellation of features. Therefore, Whole-Exome Sequencing (WES) of fetal and parental DNA was undertaken because of a strong clinical suspicion of a genetic basis for the findings. Surprisingly, WES revealed two distinct molecular diagnoses, both of which are likely to have contributed to the complex phenotype.
45X/46XX mosaicism is a well-known cause of fetal hydrops, leading us to speculate that hydropic changes, in this case, were adequately explained by this finding. However, FADS is not typically associated with 45X genotype, suggesting that the TTN mutations could have a role in this aspect of the phenotype. The TTN gene encodes the titin protein, which plays a central role in both cardiac and skeletal muscle structure and function. TTN is the largest and one of the most complex genes in the human genome.
It consists of 365 exons and, through alternate splicing, codes for a number of different isoforms of titin protein, some of which are specific to fetal life [3,4].
Titin mutations are well known to cause dominantly inherited cardiomyopathies as well as skeletal myopathies [3].
However, homozygosity or compound heterozygosity for mutations in fetal-specific isoforms of titin have been identified in babies and fetuses with multiple flexion contractures, similar to the current case. For one example, Chervinsky et al. (2018) describe an inbred pedigree with eight lethal congenital contracture syndrome cases and three intrauterine deaths caused by a TTN mutation, specifically a homozygous variant in exon 167, which is within the fetal IC isoform of titin [5]. Likewise, Fernández-Marmiesse et al. (2017) discussed a case of arthrogryposis multiplex congenita found to be secondary to a homozygous mutation in exon 197 (also part of the fetal IC isoform of titin) [6]. Although this case was not lethal, the features, including poor suction, hypotonia, and distal contractures, and bilateral humeral fractures, were severe and present at birth. Most recently, compound heterozygosity for a TTN splice site mutation in intron 213 was found to be causative of non-fatal arthrogryposis multiplex congenita in eight distinct families [7]. Considering these published cases and the predicted loss of function due to the identified variants, we considered the TTN mutations to be the cause of FADS in this fetus.
The WES findings were reviewed with the parents. They were told that the TTN mutations provided an adequate explanation for the FADS features of the baby and that the hydropic changes were likely due to the accompanying chromosome abnormality. They were advised of the 25% risk of recurrent FADS or severe, non-lethal arthrogryposis in future pregnancies. The availability of conventional prenatal diagnosis and in vitro fertilization with the pre-implantation diagnosis was explained. In a subsequent pregnancy, the patient had chorionic villus sampling at 11 weeks, which was negative for the maternal TTN mutation and positive for the paternal TTN mutation. She went on to deliver a healthy infant at 39 weeks.
This case not only adds to the literature documenting the critical role of titin in fetal muscle development but also demonstrates the importance of whole-exome sequencing in evaluating fetal demise and perinatal death. In cases of fetal demise or pregnancy termination in the context of neonatal abnormalities, the performance of whole-exome sequencing finds a causative genetic mutation in 20% of cases. Furthermore, of those cases where WES does not find a definite genetic etiology, WES will still identify possible pathogenic mutations in 45% of cases [8]. This case thus provides further support for the utility of exome sequencing in antenatal diagnosis.
Keywords
Whole exome sequencing; Perinatal death; TTN human protein; Fetal akinesia deformation sequence
Cite this article
Eisenbiegler GE, Walker AR, Brown SA. Role of whole exome sequencing in diagnosis of fetal akinesia deformation sequence with hydrops. Clin Case Rep J. 2021;2(2):1–3.
Copyright
© 2021 Brown SA. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
