Abstract
Objective: Susac Syndrome is a rare disease of unclear pathology and is often misdiagnosed as multiple sclerosis due to its variable appearance. The scope of this case report comprises presenting symptoms, diagnostic measures in terms of different treatment approaches, and possible therapeutic recommendations.
Case Presentation: A 30-year-old male patient presenting with holocephalic headaches and visual disturbances is treated with high dose prednisolone, Intravenous Immunoglobulin (IVIG), and mycophenolate mofetil after comprehensive diagnostics. An early reduction of prednisolone led to a reoccurrence of neurological impairment. Nonetheless, an added aggressive and multivalent immunotherapy with mycophenolate mofetil combined with IVIGs and prednisolone prevented further disease progression and even improved neurological symptoms to a stable condition. Patients with encephalopathy of unknown cause may therefore benefit from early ophthalmological assessment and sound threshold audiometry.
Introduction
Susac Syndrome (SS) is a rare, immunological mediated disease that affects the arterioles of the brain, retina, and inner ear, leading to vasculopathies with recurrent ischemic lesions [1] and irreversible organ damage [2]. The arising Branch Retinal Artery Occlusions (BRAO) and intracranial ischemic lesions may be detectable on retinal fluoresce in angiography and cranial magnetic resonance imaging (MRI) [3,4]. Therefore, an early diagnosis of SS is crucial for treatment initiation to govern the development of encephalopathies, visual field loss, and inner ear deafness, which characterize the symptom triad [2]. Since first described by John O. Susac in 1979 [1], over 300 cases with many different treatment regimens have been reported, but no uniform guideline has been established [5].
We demonstrate the promising approach of a SS patient with high dose prednisolone in combination with Intravenous Immunoglobulin (IVIG) and mycophenolate mofetil.
Case Presentation
A 30-year-old male patient admitted over the emergency department presented with holocephalic headaches and diffuse visual disturbances existent for eight days. In addition, he complains of recurrent paresthesia of the face and left hand. The visual disturbances are comparable to migraine symptoms with scintillating scotoma in the left field of vision and diffuse scotoma-blind spots on both eyes (left > right).
We performed Cranial non-contrast Computed Tomography (CCT) and Magnetic Resonance Imaging (MRI), with the latter revealing smaller lesions of the callosal fibers as demonstrated in (Figure 1A). Spinal MRI to expel demyelinating diseases such as encephalomyelitis disseminate was unremarkable. Visually and somatosensory evoked potentials to differentiate from Multiple Sclerosis resulted in regular latencies on both sides. Liquor diagnostics showed a pleocytosis of 18 cells with a protein of 1900mg/l. No oligoclonal bands and no further pathologic laboratory markers were evident. Ophthalmologic examination revealed 20/20 vision equivalent to full visual function but diffuse scotomas and retinal hemorrhage mainly on the left eye. In additional fluorescein angiography, peripheral vessel leakage and Branch Retinal Artery Occlusions (BRAO) were present, accentuated on the left eye to indicate retinal vasculitis and leakage of peripapillary vessels emanating from the disc (Figure 1C, Figure 1D). Evaluation by an Otolaryngologist, including audiometry, showed no hearing deficit and no sound conduction or sensation disturbance.
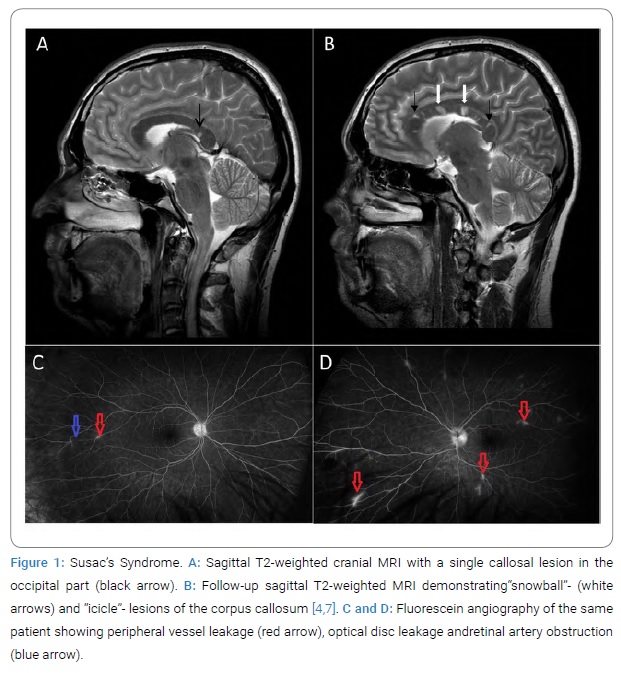
As described in the literature, the symptom triad presents not necessarily simultaneous but can occur independently at different phases of the disease. Therefore, we diagnosed Susac’s Syndrome based on the two predominant findings with pathognomonic retinal vasculitis and typical MRI findings [6].
Treatment: The patient was initially treated with intravenous high-dose cortisone therapy (1000 mg methylprednisolone) for three days with a subsequent tapered regime of initial 100 mg oral prednisolone. In a three-week follow-up, after a dose reduction to 80 mg, an assessment by the ophthalmologists revealed a decreasing vascular involvement of the retinal arteries with already improved visual acuity. No neurological deficits were present. Six weeks after initial diagnosis and a dose reduction of prednisolone to 70 mg, the patient reported deterioration with swaying vertigo, recurrent and increased cephalgia, and paresthesia of the left hand. In addition, third-party medical history revealed a psychotic syndrome with spatial cognition disorder. Ophthalmologic re-examination showed clear deterioration with increased peripheral BRAO and visual field defects (Figure 1D). Follow-up cranial MRI revealed increased callosal fiber lesions with a characteristic “icicle” and “snowball” pattern [7] (Figure 1 B).
Subsequent, two months after diagnosis, therapy escalation with a high-dose boost of methylprednisolone with 1g per day for five consecutive days and simultaneous plasmapheresis for five cycles was initiated. In addition, immunomodulatory treatment including IVIG (30 g per day) on five consecutive days as induction dose and mycophenolate mofetil was simultaneously adapted. Mycophenolate mofetil was administered two times a day with 250 mg in the first, 500 mg in the second, 750 mg in the third, and 1000 mg in the fourth week. Oral prednisolone of 100 mg was continued and carefully tapered (5 mg to 10 mg reduction every month; in the end, in smaller dose reduction as usual) over nine months. The administration of immunoglobulin (IVIGs) was given four-weekly with 30 g i.v. each on two consecutive days.
In repeated fluorescein angiography, the ophthalmologic examination revealed partial improvement of the initial vascular pathologies and cotton-wool-like foci in both eyes with fewer perivascular leakages compared to the previous month. The follow-up perimetry also revealed receding scotomas after seven months. On five months follow-up, cranial MRI showed a reduction in contrast enhancement of the cerebral lesions but no change in the lesion load. As far as the patient’s follow-up (nine-month) was recorded, no reoccurrence of any neurological symptom under this therapy with actual Prednisolon 2.5 mg was reported, and remission under the performed therapy was achieved.
Discussion
Patients suffering from Susac’s Syndrome do not always present a complete symptom triad consisting of focal neurological deficits, hearing loss, and visual field loss, making diagnosis and exclusion of differential diagnosis difficult [5,8]. Therefore, it is important to undergo cranial imaging, and if suspected, early examination of hearing and vision contributes to rapidly confirm a diagnosis and thus allows for aggressive immunosuppressive therapy at an early stage of the disease [9].
In this SS case, early administration of oral prednisolone eliminated headaches and reduced BRAO involvement. However, despite an even prolonged tapering of oral prednisolone, according to Rennebohm et al. [10]. Dose reduction from 100 mg/day to 70 mg/day after six weeks resulted in abrupt worsening of the patient’s condition from “severe” to “extremely severe” with increased neurological and visual deficits, accompanied by an increase in BRAO and MRI lesions. As described by Fox et al. [11] administration of intravenous methylprednisolone in combination with IVIG improvement of visual disturbances and cognitive dysfunction were shown. In the current literature, promising responses to immunomodulatory therapies indicated that further immunosuppressive options should be considered [3]. Thus, disease remission was achieved by adding the commonly used immunomodulator mycophenolate mofetil to the patient’s long-term treatment plan. Recent treatment approaches with natalizumab [3], and supportive regimes with acetylsalicylic acid and/or nimodipine show promising results and maybe an option in other SS cases.
Conflict of Interest
Laura van Horn, currently employed at Universitätsklinikum Hamburg Eppendorf (since December 2020). Apart from this employment there are no conflicts of interest.
Keywords
Encephalopathy; Immunomodulatory treatment; Susac syndrome; Vasculopathy
Cite this article
van Horn L, van Horn N, Eddy MT, Michels P, Röther J, Eckert B, et al. Case report of Susac Syndrome – a rare and challenging disease. Clin Case Rep J. 2021;2(4):1–4.
Copyright
© 2021 Laura van Horn. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
