Abstract
Pemphigus Vulgaris (PV) is a rare autoimmune disorder characterized by the presence of blisters on the mucous membranes and skin. Autoimmunity is a crucial complication developing in Predominantly Antibody Deficiencies (PADs), a subgroup of Primary Immunodeficiencies (PIDs). Herein, we present a case with PV who had PAD and whose disease relapsed in the maintenance period of conventional immunosuppressive treatments but was promoted to remission by high-dose Intravenous Immunoglobulin (IVIG) therapy. Thus, we aimed to create awareness for investigating PIDs in rare autoimmune bullous diseases.
Introduction
Pemphigus Vulgaris (PV) is the most common and potentially life-threatening subtype of pemphigus seen in the 4th to 6th decade of life without gender predilection [1]. The characteristic features of PV are blister formation and erosions of the skin and mucous membranes. Its incidence is approximately between 0.1–0.5/100.000. Certain ethnic groups living in the countries of the eastern Mediterranean and the Middle East are more susceptible to PV. The alleles HLA-DRB1*04:02 and HLA-DQB1*05:03 are strongly associated with PV are more common in these populations than Europeans.
Although the etiology of this disorder is generally unknown, the autoantibodies in IgG nature against desmosomal structural proteins called desmoglein (Dsg) 1 and Dsg 3 play an essential role in mucocutaneous blister formation, causing loss of cell-to-cell adhesion among the squamous epithelial cells (X). In addition, circulating autoantibodies are also found in peripheral blood of patients with PV and detected by standard serological methods such as Indirect Immunofluorescence (IIF) or Enzyme Immunoassay (EIA).
The association between PV and Predominantly Antibody Deficiency (PAD) is rarely reported until now. However, it is well known that various autoimmune diseases develop based on PADs [2]. These disorders, also called primary hypogammaglobulinemia, consist of more than 30 subtypes and 50%–60% of Primary Immunodeficiencies (PIDs) in humans. The frequencies of autoimmunity were respectively found as 23% and 26.5% in the different cohorts with PAD, including 152 and 471 patients [3,4]. This report presented a case with PV associated with PAD who had refractory to conventional treatments.
Case Presentation
A 72-year-old woman applied to our outpatient clinic because of painful ulcerous lesions in the mouth and ruptured blisters on the skin at various body parts. According to the information received from the patient, skin lesions had appeared later than the oral lesions by three months. The patient had also stated that they were fluid-filled when the lesions first appeared but burst easily in time. Her feeding had been almost completely impaired due to the lesions in the mouth. She had lost 7 kilograms in the last month. Simultaneously with the sores in the mouth, the bleeding had been developed in the whites of the eyes. Various topical and systemic antiviral and antifungal drugs had been given to the patient by different centers considering viral or fungal infection. In addition, the patient had been treated by some other physicians with high-dose oral corticosteroids without a definitive diagnosis. Despite anti-infective and immunosuppressive treatments, the lesions had become gradually widespread, and the patient’s quality of life had been impaired. The patient had not received a definitive diagnosis until she applied to our clinic.
Physical examination: Erosive exudative lesions on erythematous bases were observed on the buccal mucosa (Figure 1). Widespread erosions were also seen on upper and lower lips, palate, and tongue mucosal surfaces. Gingival mucosa was crimson in color and with an oedematous appearance. There were white patches on the tongue caused by oral thrush. Various ruptured bullous lesions were inspected on almost whole-body parts, especially under the breasts, arms, legs, and groins. Dimensions of the lesions varied from 0.5 cm x 0.5 cm being the smallest to 4 cm x 5 cm being the largest. Nikolsky’s sign was positive in the newly developed bullous lesions. In addition, there were crusted and inflamed lesions on the patient’s scalp.
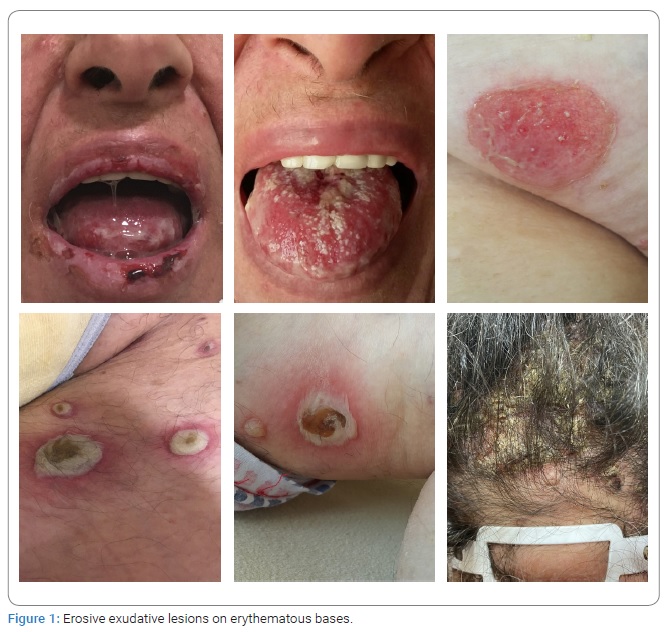
Laboratory findings: Biopsy could not be performed because the patient applied in the initial period of the Covid-19 pandemic, and therefore strict measures were taken in hospitals. At the same time, immunoglobulin G (IgG) level was below the lower limit of the reference range [(IgG: 4.8 g/L (7–16)], IgA and IgM levels were close to the lower limit of the reference range [IgA: 0.98 g/L (0.7–5), IgM: 0.34 g/L (0.3–2.9), IgE: 11.9 IU/Ml (< 87) respectively] at the time of diagnosis. When the patient’s laboratory results were searched retrospectively, it was also observed that the levels of major serum immunoglobulins four months ago had also been similar to the pattern of those of the recent measured. In addition, the percentages of lymphocyte subsets were found within the reference range. The titers of autoantibodies directed against desmoglein (Dsg) 1 and Dsg 3 in patients’ serum were 2.81 and 9.91, respectively (reference range: negative for < 1; positive for ≥ 1).
The results of routine blood test were as follows (the values measured outside the reference range are given in bold and the reference ranges are given in parenthesis): haemoglobin: 15 g/dL (12–16); haematocrit: 44.5% (35–46); white blood cell (WBC) count: 13.2∙103 cells/mm3 (4.5–11∙103); neutrophil count: 9.8∙103 cells/mm3 (2–7.8∙103); lymphocyte count: 2.6∙103 cells/mm3 (1–4∙103); platelets: 275∙103 cells/mm3 (150–400∙103); fasting glucose: 120 mg/dL (74–106); creatinine: 0.99 mg/dL (0.5–1.2); uric acid: 7.2 mg/dL (2.5–6); blood urea nitrogen (BUN): 23 mg/dL (6–20); Glomerular filtration rate (GFR): 57 mL/min/1.73 m2 (> 60); albumin: 3.7 g/dL (3.5–5); total protein: 5.9 g/dL (6–8.1); alanine aminotransferase (ALT):12 U/L (0–55); aspartate aminotransferase (AST): 14 U/L (5–34); alkaline phosphatase (ALP): 64 U/L (20–150); gamma glutamyl transferase (GGT): 12 U/L (5–36); total bilirubin: 0.78 mg/dL (0.3–1.2); lactate dehydrogenase (LDH): 222 U/L (125–243); total cholesterol: 197 mg/dL (130–200); high-density lipoprotein (HDL): 45 mg/dL (45–65); triglyceride: 110 mg/dL (55–150); C-reactive protein: 54 mg/L (0–5); erythrocyte sedimentation rate (ESR): 84 mm/hr (0–15). In addition, while serum TSH level was 3.44 mU/L (0.35–4.94), free T3 and free T4 were 1.28 ng/L (1.71–3.71) and 0.81 ng/dL (0.7–1.48), respectively. Anti-thyroid peroxidase (anti-TPO) antibody level was 2.8 IU/mL (< 5.61).
Diagnosis: According to clinical and laboratory results, the patient was diagnosed with Pemphigus Vulgaris, an autoimmune bullous disease, developed over primary antibody deficiency in the patient. When we focus on the serum levels of major immunoglobulin isotypes, the underlying immunodeficiency of the patient was categorized as probable common variable immunodeficiency (CVID) following the diagnosis algorithm suggested by Ameratunga et al. [5] and the European Society for Immunodeficiencies (ESID) criteria [5,6]. Primary hypogammaglobulinemia is the most prominent characteristic of CVID.
Treatment and clinical course: In direction with the recommendations of international boards, immunoglobulin (IVIG) treatment was started at the dose of 600 mg/kg every four weeks due to hypogammaglobulinemia [5,6]. For PV treatment, oral immunosuppressive agents were initiated as cyclosporine (CYS) 200 mg/day divided into two doses and methylprednisolone (MP) in 1 mg/kg/day in a single dose. The dose titrations of these drugs were adjusted according to clinical response. Vitamin D and Calcium supports were also given besides the specific treatments. In addition to systemic treatment, anti-ulcer and topical antifungal treatments were applied for mucosal and cutaneous lesions. The patient was taken to strict oral care.
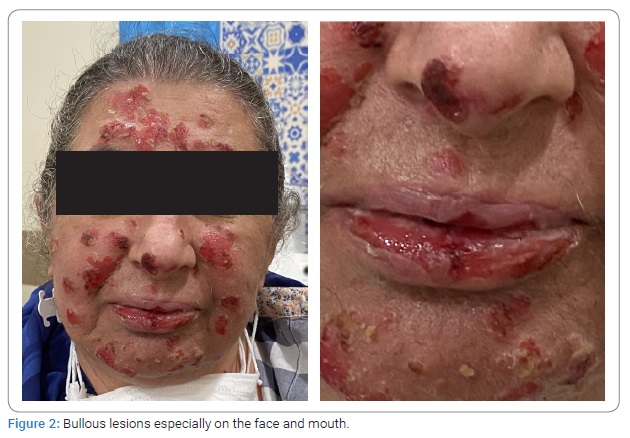
The doses of immunosuppressive drugs were gradually reduced after the clinical response was obtained and the lesions were regressed. While the patient was on maintenance treatment with 8 mg MP once a day and 50 mg CYS in two divided doses, the disease recurred on the 54th day of the treatment. Bullous lesions were observed, especially on the face and mouth (Figure 2).
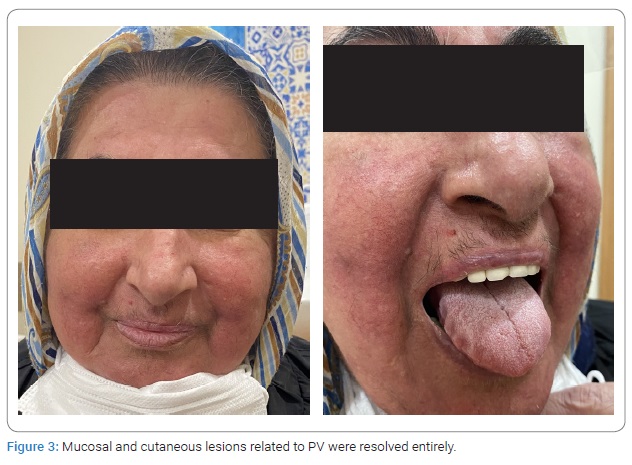
Due to metabolic complications, CYS was replaced with daily 720 mg oral mycophenolic acid (MPA) divided into two doses. The dose of MP was increased to daily 1 mg/kg as a single dose. As the first treatment period, the doses of immunosuppressive agents were gradually titrated to the maintenance doses according to clinical response. In this way, the doses of immunosuppressive agents were decreased to 360 mg/day divided into two doses for MPA, and 8 mg/day as a single dose for MP, respectively. Although a dramatic clinical improvement was achieved with this treatment regimen, new precursor lesions appeared in the mouth and body on the 104th day of the treatment. The titers of anti-Dsg1 and anti-Dsg3 autoantibodies in patients’ serum persisted as 3.58 and 5.97, respectively. Therefore, it was decided to use high-dose IVIG treatment at 2 g/kg while maintaining immunosuppressive therapy. For this purpose, the total dose of IVIG was divided into four days and given to the patient. At the end of this intervention, all mucosal and cutaneous lesions related to PV were resolved entirely (Figure 3). The treatment with MPA and MP was continued at maintenance doses without any significant side effects until the day this article was written.
During the first recurrence, hyperglycemia (fasting glucose: 285 mg/dL) and hypercholesterolemia (total cholesterol: 356 mg/dL) developed in the patient while CYS and MP were used together. Cushingoid appearance occurred due to a high dose of MP. Additionally, the levels of TSH and anti-TPO were increased to 9.29 mU/L and 122.8 IU/mL, respectively. Subcutaneous insulin treatment, besides oral formulations of atorvastatin and levothyroxine, was initiated to the patient by the endocrinologist with the diagnoses of diabetes mellitus, hyperlipidemia, and Hashimoto’s thyroiditis. The levels of fasting glucose and cholesterol were regulated with these treatments. In the last measurements, thyroid function tests were found in the normal limits. While the clinical signs of hypothyroidism have completely regressed, the cushingoid appearance partially persists.
Discussion
Common variable immunodeficiency characterized by hypogammaglobulinemia is classified into the third main group of Primary Immunodeficiencies (PIDs) according to the Phenotypical Classification table of Inborn Errors of Immunity currently suggested by the International Union of Immunological Societies (IUIS) expert committee [7]. It is well known that CVID is the most frequent symptomatic PID in adults, with an incidence of 1:50,000 to 25,000. Recurrent infections and autoimmunity are the main characteristic features of this disorder [8]. Immune cytopenia, rheumatoid arthritis, systemic lupus erythematosus, vasculitis, psoriasis, vitiligo, and immune enteropathies are common complications that develop in the course of this disease. Lymphoproliferation, granulomas, and lymphoid malignancies are the other complications in this highly heterogeneous disease.
The genetic cause can be detected in 25% of patients at most [9]. CVID includes not only antibody deficiency but also T cell defects. Because of this, severe autoimmune disorders may develop due to immune dysregulation, especially late-onset disease. In this respect, CVID is evaluated into the group of the Primary Immune Regulatory Disorders (PIRDs) tended to autoimmune diseases and have T cell and B cell defects together [8]. Tregopathies late-onset or profound combined immunodeficiencies (LoCID, P-CID) are included in the PIRD group.
Herein, we present a patient with PV developed over primary immunodeficiency. The reasons for Paraneoplastic Pemphigus (PNP) were investigated because of the elderly age and primary immunodeficiency of the patient. To reveal the common causes of PNP, lymphoid malignancies and thymoma were investigated by appropriate radiologic, biochemical, and immunologic analyses. However, we could not find any evidence related to the PNP. Thus, we excluded a vital disease that should be considered in the differential diagnosis [10].
When the literature was screened, it was seen that PV has not previously been documented to be associated with CVID. Only a woman with CVID who developed paraneoplastic pemphigus after receiving radiotherapy for relapsed non-Hodgkin’s lymphoma has been reported so far [10]. Herein, we report the first case with PV developed in a patient with CVID. Despite autoimmune skin disorders being one of the common manifestations of CVID, the fact that no such case has been reported so far may be due to the lack of awareness of PID among physicians [11]. Our primary purpose in presenting this case is to highlight that PID should be evaluated in elderly patients with autoimmune bullous disease. Otherwise, mortality and morbidity may increase with delayed diagnosis, and insufficient or inappropriate treatments may cause more devastating results.
Conflict of Interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Informed consent was obtained for this publication.
Keywords
Pemphigus vulgaris; Hypogammaglobulinemia; Autoimmune disorder
Cite this article
Musabak U, Erdogan T, Tunca M. A case of pemphigus vulgaris in a patient with primary hypogammaglobulinemia. Clin Case Rep J. 2021;2(7):1–5.
Copyright
© 2021 Tuba Erdoğan. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
