Hypokalemic Periodic Paralysis: A Case Report
* Nainanirina Sylvain;
Solohery Jean Noël Ratsimbazafy;
Ainasalohimanana Marie Ymelda Randrianarisoa;
Solofo Andriambelo Ralamboson;
Hanta Marie Danielle Vololontiana;
-
* Nainanirina Sylvain: USFR Internal Medicine, CENHOSOA of Antananarivo, Madagascar.
-
Solohery Jean Noël Ratsimbazafy: USFR Internal Medicine, CENHOSOA of Antananarivo, Madagascar.
-
Ainasalohimanana Marie Ymelda Randrianarisoa: USFR Internal Medicine, CHUJRB of Antananarivo, Madagascar.
-
Solofo Andriambelo Ralamboson: USFR Internal Medicine, CENHOSOA of Antananarivo, Madagascar.
-
Hanta Marie Danielle Vololontiana: USFR Internal Medicine, CHUJRB of Antananarivo, Madagascar.
Abstract
Introduction: Hypokalemic periodic paralysis (HPP) or Westphall’s disease is a genetic channelopathy with autosomal dominant transmission at the origin of transfer hypokalemia. We report the first observation of a Malagasy patient in Madagascar to incite physicians to evoke it in front of an association of hypokalemia and a neurological deficit.
Case Presentation: This was a 45-year-old woman, hypertensive, admitted to the emergency room for a sudden motor deficit of the four limbs, predominantly right, evolving since the day of her admission. She had a history of a similar episode in the left lower limb but no similar family history. The physical examination showed asymmetric tetraparesis. There was no sensory deficit. The examination of the cranial pairs was normal. Osteotendinous and cutaneous-plantar reflexes were absent. Thyroid workup, muscle enzymes (creatine phosphokinase, lactate dehydrogenase), and brain scan were normal. However, she had hypokalemia with electrical repercussions such as prolonged QT. The diagnosis of hypokalemic periodic paralysis was evoked. Potassium supplementation by intravenous infusion was instituted. The evolution was marked by a complete recovery of muscle strength within 24 hours, concomitant with the normalization of the kalemia.
Conclusion: Hypokalemic periodic paralysis is a rare disease but should be known by physicians. The diagnosis is made based on hypokalemia not explained by secondary causes associated with a neuromuscular deficit and confirmed by genetic testing. Treatment is based on the correction of hypokalemia and avoidance of triggering factors.
Introduction
Hypokalemic periodic paralysis (HPP) or Westphall’s disease is a genetically inherited autosomal dominant channelopathy causing transfer hypokalemia [1]. It is a rare disease but the most frequent of the periodic paralysis, its precise prevalence is not known, but it would be about 1/100000 [2,3]. We report this first case to encourage physicians to evoke it before the association of paralysis and hypokalemia.
Case Presentation
This was a 45-year-old hypertensive woman on dual therapy (Perindopril 10 mg/d and Amlodipine 10 mg/d) and a normal diet. She was admitted to the emergency room for a sudden deficit of the right limbs in an apyretic context, without any notion of hypertensive attack, occurring after an unusual physical exercise. A stroke diagnosis was evoked at first, but the cerebral scanner was normal.
She reported a previous episode in the left lower limb one week before. There was no similar family history.
The hemodynamic status was stable and apyretic on physical examination with blood pressure at 130/80 mmHg, heart rate at 80 beats/min, respiratory rate at 16 cycles/min, oxygen saturation (SpO2) at 96% on room air, and apyrexia at 36.7°C. The neurological examination showed asymmetric tetraparesis with flaccid paralysis of the right lower limb (muscle score of 0/5), mild paresis of the left lower limb at 4/5, and predominantly distal paresis of the upper limbs with a muscle score of 4/5. There was no sensory deficit. The examination of the cranial pairs was normal. Osteotendinous and cutaneous-plantar reflexes were absent. The rest of the clinical respiratory and cardiovascular examination was unremarkable.
On biological examination, severe hypokalemia of 2.2 mmol/l was observed. Creatinine (89 umol/l), uremia (4.4 mmol/l), initial determinations with controls of LDH and CPK enzymes, and thyroid workup were normal. Genetic testing was not feasible in our country. The urinary ionogram revealed a kaliuresis of 14 mmol/l.
The electrocardiogram (ECG) showed a sinus rhythm with a QT prolongation with corrected QT 753 ms related to hypokalemia (Figure).
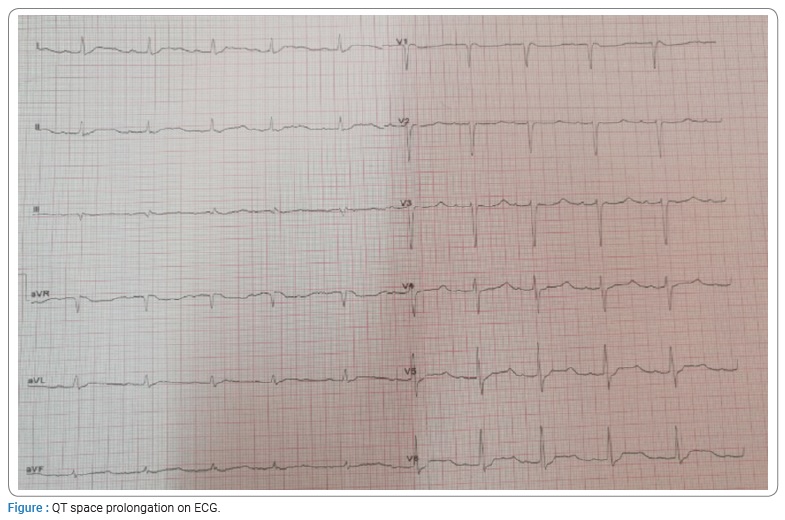
The diagnosis of hypokalemic paralysis was evoked.
Correction of hypokalemia by intravenous infusion of 2 g of KCl (potassium chloride) over eight hours was instituted. The evolution was marked clinically by the progressive recovery of muscle strength and the complete disappearance of the deficit at the 24th hour. Concomitant with the normalization of the kalemia and paraclinical, the kalemia was 3.7 mmol/l with a kaliuresis of 60 mmol/24h, and the ECG had returned normal.
Discussion
PPH is a rare pathology whose pathophysiology is still poorly understood. It involves genetic mutations occurring in the genes of the α subunit of a voltage-dependent L-type calcium channel (CACNA1S) in 70% of cases or the genes of the α subunit of the muscle sodium channel (SCN4A) in 10% [4]. This pathology presents triggering factors such as the intake of food rich in carbohydrates or salts, alcohol, strong emotion, and rest after intense or unusual physical exercise, as in the case of our patient [4]. It generally apart during the second decade and is characterized, in its most frequent form (75%) by the occurrence of iterative episodes of tetraplegia or flaccid paralysis lasting from a few hours to a few days, sparing the respiratory, oculomotor, and bulbar muscles and the heart [4,5]. In our case, it was predominantly right tetraparesis with flaccid paralysis of the right lower limb. In the face of this picture of sudden neurological deficit, stroke should be ruled out in the first instance by brain imaging. The diagnosis of PPH is clinical and biological but can be confirmed by genetic analysis, the negativity of which does not eliminate the diagnosis [6,7].
Hypokalemia is most often between 0.9 mmol/l and 3 mmol/l [4,5], which is the case for our patients. The ECG more frequently shows the classical signs of hypokalemia, namely ST-segment depression, T-wave depression or inversion, increased U-wave amplitude, and QT prolongation, as in our patient.
Although rare, the most formidable complication of a PPH attack is the occurrence of a threatening intraventricular conduction disorder of ventricular fibrillation [8].
Treatment consists of careful correction of hypokalemia, not exceeding a dose of 10 mmol/hr, as too rapid correction exposes the risk of hyperkalemia [9].
Thus, regular surveillance by physical examination and monitoring of kalemia is essential.
In addition, therapeutic education focused on avoiding triggers is important to prevent the occurrence of seizures.
Conclusion
PPH is a rare disease, but the physician should know it. The diagnosis is made based on hypokalemia not explained by secondary causes associated with a neuromuscular deficit and confirmed by genetic testing. Treatment is based on the correction of hypokalemia and avoidance of triggering factors.
Acknowledgments
- Dr. Ida Marie Rahantamalala, USFR Internal Medicine, CHU Anosiala Antananarivo, Madagascar.
- Dr. Solofo Andriambelo Ralamboson, USFR Internal Medicine, CENHOSOA Antananarivo, Madagascar.
- USFR Internal Medicine, CHUJRB of Antananarivo, Madagascar.
Authors’ contributions: All authors contributed to the conduct and completion of this work.
Conflict of Interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Informed consent was obtained for this publication.
References
- Goichot B. Hypokaliémies génétiques. Rev Med Interne. 2001;22:255–642.
- Fontaine B. Periodic paralysis. Adv Genet. 2008;63:3–23.
- Kantola IM, Tarssanen LT. Familial hypokalaemic periodic paralysis in Finland. J Neurol Neurosurg Psychiatry 1992;55(4):322–324.
- Ke Q, Luo B, Qi M, Du Y, Wu W. Gender differences in penetrance and phenotype in hypokalemic periodic paralysis. Muscle Nerve. 2013;47(1):41–45.
- Bourdain F, Fontaine B. Canaux ioniques dépendants du voltage et maladies neuromusculaires. Encycl Med Chir (Elsevier, Paris), Neurologie. 2005;17:001–020.
- Stapleton LJ. Hypokalaemia periodic paralysis. Scott Med J. 2018;63(1):28–31.
- Levitt JO. Practical aspects in the management of hypokalemic periodic paralysis. J Transl Med. 2008;6:18.
- Boccalandro C, Lopez L, Boccalandro F, Lavis V. Electrocardiographic changes in thyrotoxic periodic paralysis. Am J Card. 2009;91:775–777.
- Burtey S, Vacher-Coponat H, Berland Y, Dussol B. [About two cases of hypokaliemic periodic paralysis]. Nephrol Ther. 2006;2:379–386.
Keywords
Madagascar; Hypokalemia; Periodic paralysis
Cite this article
Nainanirina S, Ratsimbazafy SJN, Randrianarisoa AMY, Ralamboson SA, Vololontiana HMD. Hypokalemic periodic paralysis: a case report. Clin Case Rep J. 2022;3(4):1–3.
Copyright
© 2022 Nainanirina Sylvain. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
