Retinitis Pigmentosa 45: A Case Report
* Rita Costa Basto;
Sara Pereira;
Renato Barbosa;
Ana Rita Viana;
Rita Gonçalves;
Carla Teixeira;
-
* Rita Costa Basto: Department of Allergy and Clinical Immunology, San Giovanni di Dio Hospital, Florence, Italy.
-
Sara Pereira: Department of Allergy and Clinical Immunology, San Giovanni di Dio Hospital, Florence, Italy.
-
Renato Barbosa: Department of Allergy and Clinical Immunology, San Giovanni di Dio Hospital, Florence, Italy.
-
Ana Rita Viana: Department of Allergy and Clinical Immunology, San Giovanni di Dio Hospital, Florence, Italy.
-
Rita Gonçalves: Department of Allergy and Clinical Immunology, San Giovanni di Dio Hospital, Florence, Italy.
-
Carla Teixeira: Department of Allergy and Clinical Immunology, San Giovanni di Dio Hospital, Florence, Italy.
Abstract
Clinical case of a 36-year-old man with symptomatic peripheral visual field loss. Electroretinography showed severely reduced rod function. The DNA test revealed homozygous deletion mutation in CNGB1, associated with Retinitis Pigmentosa type 45, characterized by an early onset with a rather slow progression of the disease.
Introduction
Retinitis pigmentosa (RP) is inherited retinal dystrophy affecting 1 in 3000-5000 patients [1,2]. The term is used for a group of genetically and clinically heterogeneous disorders with diffuse affection of photoreceptors and retinal pigment epithelium (RPE). In comparison to cone function, Rod function is usually impaired earlier and more severely, so the term rod-cone dystrophy is also applied [3].
The typical evolution is the progressive degeneration and death of rod photoreceptors, resulting in loss of night vision (nyctalopia) and a gradual constriction of the visual field. In addition, ophthalmic evaluation classically reveals bone spicule formation in the peripheral retina, blood vessel attenuation, and optic disc pallor. RP is also associated with posterior subcapsular cataracts in 39% to 72% of patients, high myopia, astigmatism, keratoconus, and mild hearing loss in 30% of patients [3].
RP is genetically extremely heterogeneous, with more than 50 disease-causing genes currently associated with this condition [4]. Fifteen to 20% of RP cases are autosomal dominant (AD); 15% are autosomal recessive (AR); 7% are X-linked; 43% are sporadic or simplex cases, most of which are likely to be AR; 15% are unknown, and rarely RP may be caused by mutations in mitochondrial DNA [5,6].
Two major genes are responsible for AR inheritance, USH2A, and EYS. Mutations in the rod cycling nucleotide-gated channel B1 (CNGB1) gene cause AR RP type 45 (RP45) and account for approximately 4% of AR inheritance [7]. CNGB1 gene, found in chromosome 16q21 [8], encodes cyclic nucleotide-gated (CNG) beta-1 subunits and two soluble glutamic-acid rich proteins (GARPs) [9]. CNG channel is a heterotetramer of three alfa subunits (CNGA1) and one beta-subunit (CNGB1) [5]. CNG is a nonselective cation channel known to be important for both visual and olfactory transduction pathways [10].
Patients with RP45 typically describe night blindness from childhood and a later-onset and slowly progressive loss of cone function and constriction of visual fields, leading to an RP diagnosis at around 30 years of age and legal blindness by 60 years of age [7].
Case Presentation
A 36-year-old Portuguese man of a non-consanguineous family with RP was studied. The pedigree demonstrated only one affected individual.
Symptomatic peripheral visual field loss occurred at the age of 34 years. The patient did not complain of night blindness or photophobia.
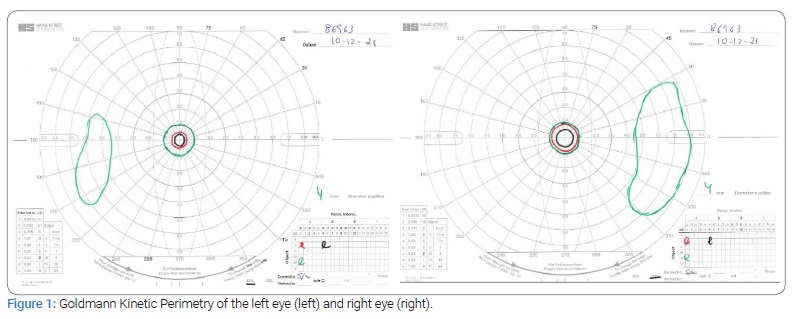
During his first ophthalmic examination (at 35 years), the best-corrected visual acuity was 0.1 logMAR (Snellen 8/10) in the right eye and 0.0 logMAR (Snellen 10/10) in the left eye. In addition, there was sparing of the central ten degrees and a temporal islet of the visual field in both eyes, revealed by Goldmann Kinetic perimetry (Figure 1).
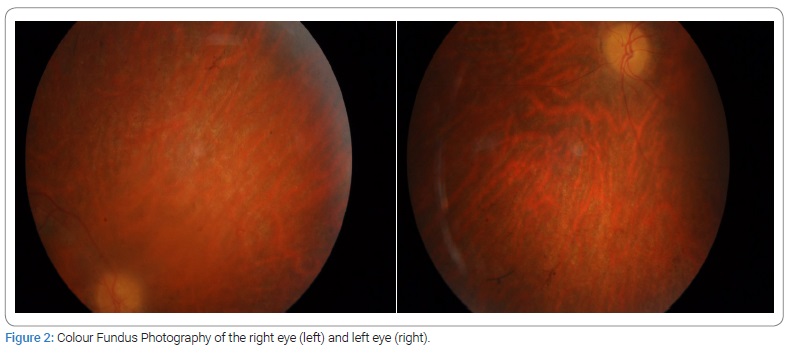
Anterior segment biomicroscopy showed a bilateral incipient subcapsular cataract. In addition, mydriatic fundus examination revealed arteriolar attenuation, optic disc pallor, RPE atrophy, and mid-peripheral intraretinal spicule-shaped pigmentary deposits (Figure 2).
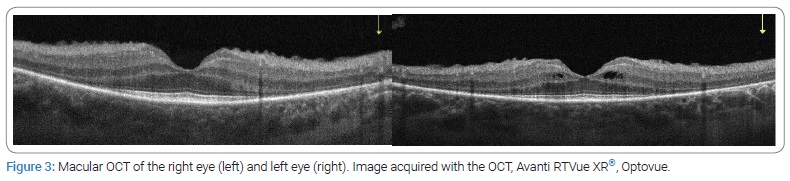
Optical Coherence Tomography (OCT) revealed a bilateral loss of the ellipsoid (ISe) band and photoreceptor layer, only preserved subfoveally and intraretinal cysts in the left eye (Figure 3).
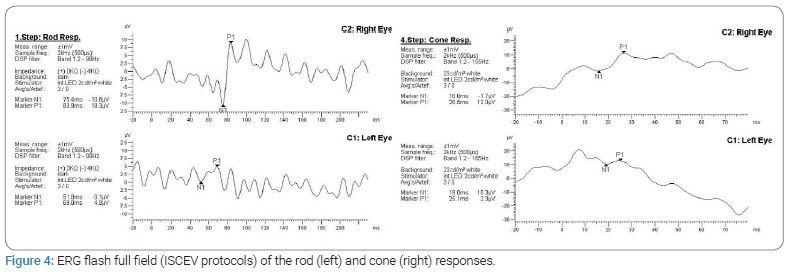
Full-field Electroretinography (ERG) showed profoundly attenuated rod-specific responses (dark-adapted) with subnormal and delayed cone responses (Figure 4).
DNA samples obtained from the patient were sequenced using target exome-based next-generation sequencing. To identify the molecular basis of RP in this patient, we screened a panel of 307 genes associated with posterior segment ocular diseases by targeted next-generation sequencing and direct confirmatory sequencing. We found a homozygous mutation of the CNGB1 gene, segregating with the retinitis pigmentosa phenotype. In addition, we identified homozygous deletion mutation in CNGB1: c3150del p. (Phe1051Leufs*12). These mutations are the potential to cause disease and are classified as a “likely pathogenic” variant based on ACMG guidelines [11].
Discussion
Electroretinographic responses and Goldmann kinetic perimetry are both good measures of the remaining functioning retina in nonsyndromic RP, irrespective of inheritance models and dystrophic patterns [12]. Visual field loss has a great impact on a patient’s quality of life and is an important indicator of disease progression. In our case, the patient maintains a good vision. However, losing a mostly peripheral visual field impacts his daily activities.
The prevalence of cystoid macular edema (CME) in cases of retinitis pigmentosa is demonstrated in some studies [13,14]. Its pathogenesis is not clearly understood. The dysfunction of the outer blood-retinal barrier, and the consequent increase in vascular permeability, may be responsible for the fluid leakage through the RPE. RP-associated CME may show only mild or no angiographic evidence of leakage or pooling of the dye, making OCT the best choice for its diagnosis.
The involvement of CNGB1 in RP was first reported in 2001 using homozygosity mapping to identify a homozygous variant (c.2978G > T). To date, only 13 pathogenic CNGB1 variants have been reported.
RP45 seems to have an early onset with a rather slow progression, but a genotype-phenotype correlation has yet to be established [15–17]. Consistent with the clinical characterization of RP, the CNGB1-RP patients maintained a central area of relatively preserved visual function and acuity [18–21].
The expression of CNGB1 in the retina is restricted to rod photoreceptors. Though, the observed cone dysfunction evident in ERG of our case is likely consequent upon outer retinal degeneration [9].
Conclusion
Our case confirms that RP associated with variants in CNGB1 has a good prognosis for central vision despite the early onset of peripheral visual field loss. In addition, the patient does not report characteristic symptoms of night blindness. The good visual prognosis is reflected by preserved central macular thickness and morphology of the ISe band subfoveally and photoreceptor layer.
The causative gene needs to be identified with the advent of clinical trials for inherited retinal dystrophies. Molecular identification permits the diagnosis of the Retinitis pigmentosa (RP) subtype, improving the patient follow-up and the prediction of disease course. Gene identification is also necessary for the development of gene therapy.
Compliance with Ethical Standards
Ethical approval: The patient and his family have given written informed consent as they authorized this study. The study protocol follows the 2013 Helsinki declaration and its later amendments.
Conflict of Interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Informed consent was obtained for this publication.
References
- Newton F, Megaw R. Mechanisms of photoreceptor death in retinitis pigmentosa. Genes (Basel). 2020;11(10):1120.
- Jauregui R, Park KS, Duong JK, Mahajan VB, Tsang SH. Quantitative progression of retinitis pigmentosa by optical coherence tomography angiography. Nature. 2018;8:13130.
- Seeliger M, Kretschmann U, Apfelstedt-Sylla E, Rüther K, Zrenner E. Multifocal electroretinography in retinitis pigmentosa. Am J Ophthalmol. 1998;125(2):214–226.
- Bocquet B, al Dain Marzouka N, Hebrard M, Manes G, Sénéchal A, Meunier I, et al. Homozygosity mapping in autosomal recessive retinitis pigmentosa families detects novel mutations. Mol Vis. 2013;19:2487–2500.
- Hull S, Attanasio M, Arno G, Carss K, Robson AG, Thompson DA, et al. Clinical characterization of CNGB1-related autosomal recessive retinitis pigmentosa. JAMA Ophthalmol. 2017;135(2):137–144.
- Nishiguchi KM, Tearle RG, Liu YP, Oh EC, Miyake N, Benaglio P, et al. Whole genome sequencing in patients with retinitis pigmentosa reveals pathogenic DNA structural changes and NEK2 as a new disease gene. Proc Natl Acad Sci U S A. 2013;110(40):16139–16144.
- Petersen-Jones SM, Occelli LM, Winkler PA, Lee W, Sparrow JR, Tsukikawa M, et al. Patients and animal models of CNGβ1-deficient retinitis pigmentosa support gene augmentation approach. J Clin Invest. 2018;128(1):190–206.
- RetNet. Genes and mapped loci causing retinal diseases [Internet]. USA: RetNet; 2021.
- Ba-Abbad R, Holder GE, Robson AG, Neveu MM, Waseem N, Arno G, et al. Isolated rod dysfunction associated with a novel genotype of CNGB1. American Journal Of Ophthalmology. 2019;14(83–86).
- Bareil C, Hamel CP, Delague V, Arnaud B, Demaille J, Claustres M. Segregation of a mutation in CNGB1 encoding the beta-subunit of the rod cGMP-gated channel in a family with autosomal recessive retinitis pigmentosa. Hum Genet. 2001;108(4):328–334.
- ACMG. Practice Guidelines[Internet]. Maryland: American college of medical genetics and genomics; 2021.
- Iannaccone A, Rispoli E, Vingolo EM, Onori P, Steindl K, Rispoli D, et al. Correlation between Goldmann perimetry and maximal electroretinogram response in retinitis pigmentosa. Doc Ophthalmol. 1995;90(2):129–142.
- Hirakawa H, Iijima H, Gohdo T, Tsukahara S. Optical coherence tomography of cystoid macular edema associated with retinitis pigmentosa. Am J Ophthalmol. 1999;128(2):185–191.
- Chung H, Hwang JU, Kim JG, Yoon YH. Optical coherence tomography in the diagnosis and monitoring of cystoid macular edema in patients with retinitis pigmentosa. Retina. 2006;26(8):922–927.
- Fradin M, Colin E, Hannouche-Bared D, Audo I, Sahel JA, Biskup S, et al. Run of homozygosity analysis reveals a novel nonsense variant of the CNGB1 gene involved in retinitis pigmentosa 45. Ophthalmic Genet. 2016;37(3):357–359.
- Banerjee S, Yao J, Zhang X, Niu J, Chen Z. Next generation sequencing identified novel heterozygous nonsense mutation in CNGB1 gene associated with retinitis pigmentosa in a Chinese patient. Oncotarget. 2017;8(51):88345–88350.
- Kondo H, Qin M, Mizota A, Kondo M, Hayashi H, Hayashi K, et al. A homozygosity-based search for mutations in patients with autosomal recessive retinitis pigmentosa, using microsatellite markers. Invest Ophthalmol Vis Sci. 2004;45(12):4433–4439.
- Manlong X, Zhai Y, MacDonald IM. Visual field progression in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2020;61(6):56.
- Seo JH, Yu HG, Lee BJ. Assessment of functional vision score and vision-specific quality of life in individuals with retinitis pigmentosa. Korean J Ophthalmol. 2009;23(3):164–168.
- Ross DF, Fishman GA, Gilbert LD, Anderson RJ. Variability of visual field measurements in normal subjects and patients with retinitis pigmentosa. Arch Ophthalmol. 1984;102(7):1004–1010.
- Sugawara T, Hagiwara A, Hiramatsu A, Ogata K, Mitamura Y, Yamamoto S. Relationship between peripheral visual field loss and vision-related quality of life in patients with retinitis pigmentosa. Eye (Lond). 2010;24(4):535–539.
Keywords
Retinal dystrophy; Night blindness; Rod photoreceptors; Homozygosity; CNGB1 gene
Cite this article
Basto RC, Pereira S, Barbosa R, Viana AR, Gonçalves R, Teixeira C. Retinitis Pigmentosa 45: a case report. Clin Case Rep J. 2022;3(4):1–4.
Copyright
2022 Rita Costa Basto. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
