Pulmonary Light Chain Disease: An Unusual Presentation in a 54-Year-Old Man with Pulmonary Cysts and Granulomatous Inflammation with Localized Vasculitis
Samantha Jakuboski;
Imran Uraizee;
Paul Camren;
Girish Venkataraman;
Daniel Dilling;
* Vijayalakshmi Ananthanarayanan;
-
Samantha Jakuboski: Department of Pathology, Stritch School of Medicine, Loyola University Chicago, IL, USA.
-
Imran Uraizee: Department of Pathology, Loyola University Medical Center, IL, USA.
-
Paul Camren: Department of Radiology, Loyola University Medical Center, IL, USA.
-
Girish Venkataraman: Department of Pathology, University of Chicago, IL, USA.
-
Daniel Dilling: Division of Pulmonary and Critical Care Medicine, Loyola University Medical Center, IL, USA.
-
* Vijayalakshmi Ananthanarayanan: Department of Pathology, Loyola University Medical Center, IL, USA; Division of Pulmonary and Critical Care Medicine, Loyola University Medical Center, IL, USA.
Abstract
A 54-year-old asymptomatic male with a ten-year history of a left lung nodule presented for a surgical lung biopsy following a chest scan for increased nodule size and newly identified perivascular cysts. Histopathological examination showed necrotizing, granulomatous inflammation with neutrophils, eosinophils, lymphocytes, and plasma cells, with surrounding vasculitis; another section showed accumulation of amorphous, eosinophilic material without granulomas or vasculitis. This case was initially broadly classified as cystic lung disease. However, the patient was ultimately diagnosed with pulmonary Light Chain Deposition Disease (LCDD) secondary to low-grade lymphoma. LCDD of the lung, known as pulmonary LCDD (PLCDD), is rare; since the identification of PLCDD in 1988, around 50 cases of the disease have been documented. For patients presenting with an accumulation of clonal light chains and vasculitis, an underlying low-grade lymphoma should thus be considered.
Case Presentation
A 54-year-old male presented for pulmonary consultation following a lung wedge resection performed six months prior at an outside consultation for a slowly growing lung nodule. Since his wedge resection, the patient endorsed a mild cough but denied shortness of breath, hemoptysis, or increased sputum production. He reported good physical endurance, riding 50–100 miles per week on his bike. On physical exam, the patient weighed 94.3 kg with a height of 175.3 cm. Auscultation revealed bilateral clear breath sounds, with no wheezing, rales, rhonchi, dullness to percussion, or chest tenderness. Vital signs were within normal limits, with a blood pressure of 138/84 mmHg, heart rate of 88 beats/minute, and 99% oxygen saturation on room air. Recent pulmonary function tests revealed an FVC of 4.26 L (92% predicted), FEV1 of 3.29 L (98% predicted), FEV1/FVC of 77%, and a DLCO of 36 ml/min/mmHg (137% predicted). Genetic testing for Birt-Hogg-Dubé syndrome and tuberous sclerosis susceptibility genes (FLCN, TSC1, and TSC2, respectively) was negative.
Post-surgical biopsy high-resolution computed tomography (HRCT) (Figure 1) showed many bilateral, thin-walled, heterogeneously distributed perivascular cysts, some along pleural surfaces or lung fissures, with a propensity for lower lung fields. The cysts were increased in number compared to prior imaging.
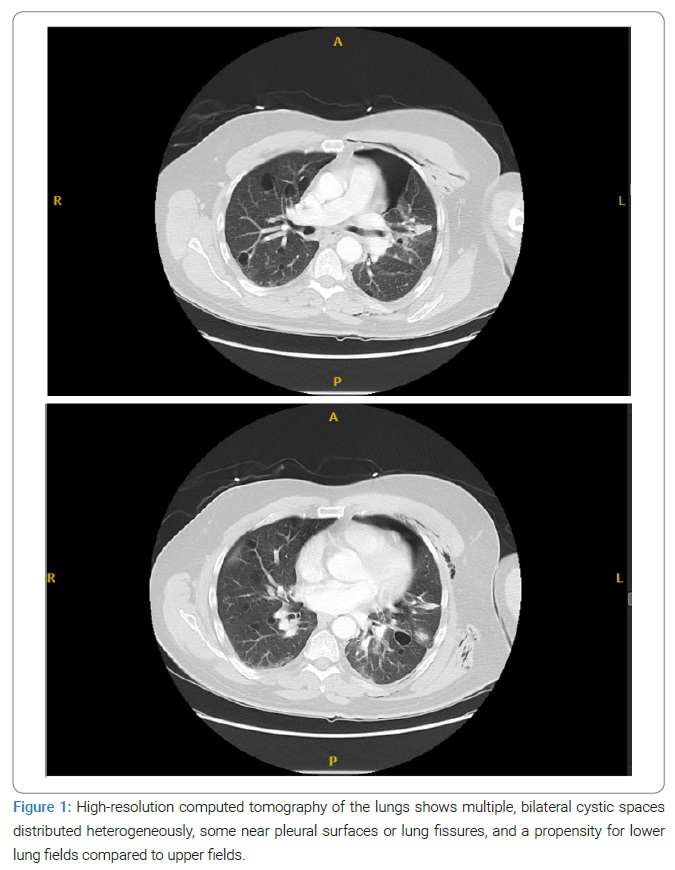
Other findings included emphysematous changes, a new left simple pneumothorax over the left lung apex, an ill-defined opacity in the lateral left upper lobe (likely related to recent post-surgical changes), and focal consolidation in the medial and inferior left lower lobe with a degree of volume loss. Extensive soft tissue emphysema surrounding the left hemithorax was appreciated.
Two wedge biopsies were taken. Histologic examination of one of the wedge resections from the left lower lung lobe showed a necrotic, infarcted, well-demarcated nodule rimmed by granulomatous inflammation composed of mixed infiltrate of lymphocytes, histiocytes, eosinophils, multinucleated giant cells, and plasma cells, including admixed multinucleated plasma cells (Figure 2). At the peripheral interface of the nodule, localized vasculitis with brisk, mural inflammatory infiltrates with eosinophils resulting in vascular obliteration of medium-to-large sized vessels was noted, raising consideration of vasculitides such as granulomatosis with polyangiitis. The background lung parenchyma did not show evidence of vasculitis; many non-necrotizing granulomas with follicular bronchitis and bronchiolitis were noted. AFB and GMS stains were negative for acid-fast and fungal organisms, respectively.
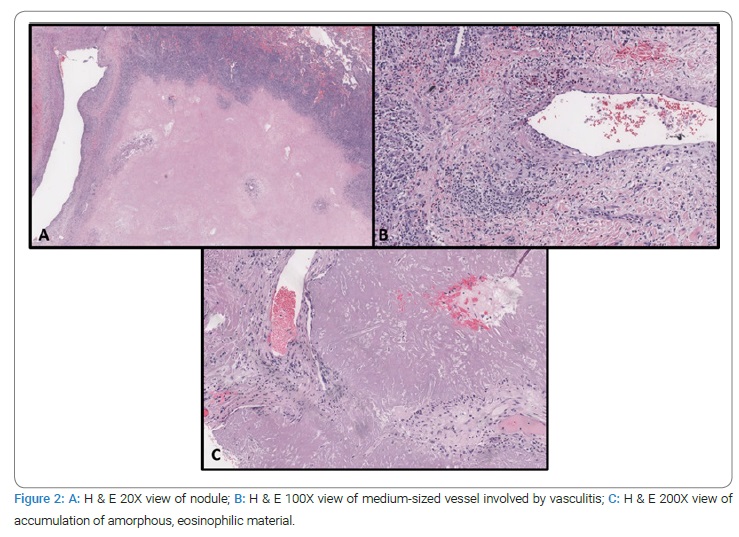
The second wedge resection showed an accumulation of granular, amorphous, eosinophilic material with punctate calcifications surrounded by a rim of histiocytes and foamy, multinucleated giant cells (Figure 2C).
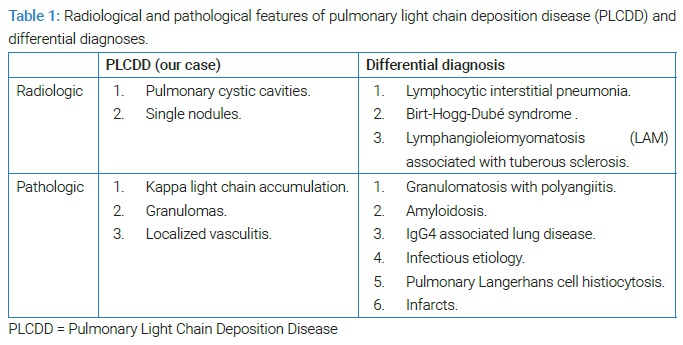
These features led to the consideration of amyloidosis, IgG4-associated lung disease, pulmonary Langerhans cell histiocytosis, pulmonary light chain deposition disease, infection, and pulmonary infarction in the differential diagnosis (Table 1).
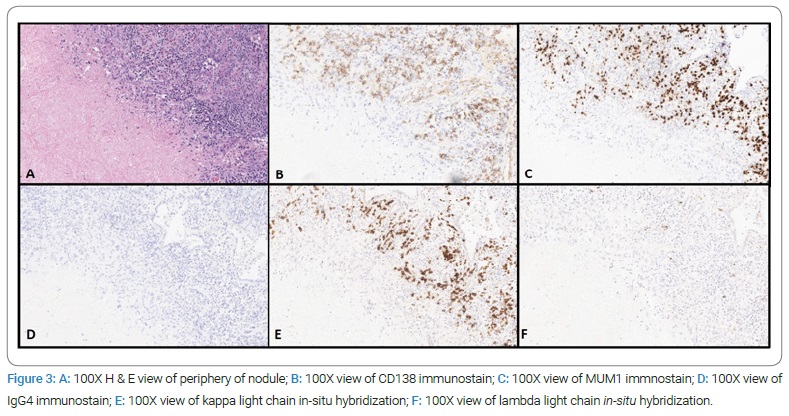
Immunohistochemical stains for CD20, MUM1, CD138, IgG4, kappa, lambda light chain in-situ hybridization, and Congo red stains were performed on a representative block. CD20 showed scattered B cells. There was no increase in IgG4-positive plasma cells. Congo red stain for amyloid was negative. Of note, in situ light chain hybridization analysis showed the plasma cells surrounding the necrotic nodule to be clonally kappa-restricted, raising the possibility of an underlying localized or systemic B-cell lymphoproliferative disorder. Representative images of the nodule interface and special stains are shown in (Figure 3). Formalin-fixed, paraffin-embedded blocks were sent to an outside institution for Liquid Chromatography-Tandem Mass Spectrometry (LC/MS) light chain analysis, which confirmed extracellular deposits to be comprised of kappa immunoglobulin light chains and the absence of proteomic features of amyloid.
Discussion
Our patient presented with an enlarging lung nodule and parenchymal cysts, prompting further investigation and a wedge biopsy. He was ultimately diagnosed with pulmonary light chain deposition disease (LCDD) secondary to low-grade lymphoma (suggested extranodal marginal zone lymphoma). LCDD is a non-amyloid monoclonal light chain disorder affecting middle-aged adults, characterized by extracellular accumulation of immunoglobulin (Ig), mainly IgG kappa light chain, in tissues. The kidneys are the most common sites of LCDD, with extrarenal sites including the lung, liver, and heart [1,2]. Though clinically similar to amyloidosis, LCDD has key differences: the presence of granular rather than fibrillar deposits, negative Congo red staining due to the lack of the β-pleated light chain configuration, and the lack of apple-green birefringence under polarized light [2,3]. LCDD of the lung, known as pulmonary LCDD (PLCDD), is rare. Since the identification of PLCDD in 1988, around 50 cases of the disease have been documented [4,5]. 75% of LCDD cases occur in conjunction with a lymphoproliferative disease or multiple myeloma [1,2]. While some patients are asymptomatic, others may present with cough, dyspnea, chest discomfort, night sweats, and weight loss [5–7].
Chest CT, though not diagnostic in PLCDD, may reveal pulmonary cystic cavities; multiple, singular, bilateral nodules, ranging in size from 1 mm–5 mm; and/or fibrosis of the interstitium or parenchyma [4–7]. The most consistent finding is multiple thin-walled cysts throughout the lungs often with a basilar predominance. Pulmonary vessels along the periphery of the cysts may cause focal cyst wall thickening or nodularity. Over time, the cysts may enlarge. Scattered small lung nodules and reticular opacities have also been described. The differential diagnosis for cystic lung disease with numerous intraparenchymal cysts includes lymphocytic interstitial pneumonia (LIP), Birt-Hogg-Dubé syndrome, sporadic lymphangioleiomyomatosis (LAM), and LAM associated with tuberous sclerosis (Table 1).
PLCDD can present with a combination of amorphous eosinophilic material comprised of light chains (kappa and/or lambda), lymphocytic infiltrates of B and T cells, collagenous tissue, multinucleated giant cells with or without granulomas, and hyalinizing granulomas surrounded by whorled lamellar collagen deposited in the alveolar septa, space, and blood vessel walls [6,7]. Kappa restriction is often identified on immunohistochemical staining, with the kappa to lambda light chain ratio reaching 10:1 [7]. On electron microscopy, uranyl acetate and lead citrate can be used to stain for the light chain accumulations.
Bhargava and colleagues classify the histologic patterns of PLCDD as either diffuse or nodular [7]. The diffuse form presents with a preserved pulmonary architecture and thickened basement membrane [7]. The second form, nodular, is characterized by the presence of one or more nodules in a relatively healthy patient [7]. The presence of granulomas has historically made the diagnosis of PLCDD difficult [7]. Nodules can be mistaken for infectious granulomatous disease, as was initially suspected in our patient [7]. The absence of central necrosis and infiltration of neutrophils in infectious granulomatous disease can help differentiate the two diseases [7]. Other differentials to keep in mind with the nodular form include pulmonary hyalinizing granuloma and nodules of either sarcoidosis or silica exposure [7]. Since 50% of PLCDD patients will have a plasma cell dyscrasia or a lymphoproliferative disorder, a correct and timely diagnosis of PLCDD is important to address the underlying hematological disorder [7]. The association between LCDD and low-grade B-cell lymphoma has been reported in select cases of patients [8,9]. Cysts represent a localized plasma cell response with the deposition of light chains. We postulate this accumulation creates a hypercoagulable state, blocking blood vessels and increasing the likelihood of infarct. The subsequent granulomatous response to the infarct then creates a nodule. While our patient has both cysts and nodules, the case is unusual in that the nodule presented before the cysts, reversing the progression mentioned above.
Another unusual finding that our patient presented with is localized vasculitis. Literature on the association between vasculitis and LCDD is sparse, but some case reports have reported a similar correlation between vasculitis and monoclonal immunoglobulin deposition disease. An early case report described two patients with vasculopathies of the kidneys and extremities with a monoclonal lambda light chain component. One patient also had crystalline deposits of lambda light chain in small vessels of organs, including the lung and alveoli [10]. Bone marrow examination showed hypercellularity and plasmacytosis, suggestive of a plasma cell dyscrasia [10]. Another case report described a patient who presented with a necrotizing vasculitis and was eventually diagnosed with gamma-heavy chain disease [11].
After the clinical consultation, review of pathology, and multidisciplinary discussion, a recommendation was made for the patient to follow-up with the hematology-oncology team. Unfortunately, the patient was lost to follow-up; no systemic therapy was instituted while the patient was under our care.
Conclusion
This was an interesting and diagnostically challenging case for several reasons:
- First, the indolent nature of this case, with the patient asymptomatic and undiagnosed for over ten years since the nodule was first observed.
- Second, the distinct histologic findings of necrotizing, granulomatous inflammation with localized vasculitis in one wedge biopsy and amorphous light chain deposits in the second wedge biopsy.
- Lastly, the scant hematolymphoid cells on histologic examination added to the challenge of diagnosing an underlying low-grade lymphoma. For patients presenting with an accumulation of clonal light chains and vasculitis, an underlying low-grade lymphoma should be considered.
Acknowledgments
Funding: This work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Ethics approval: This study was conducted in accordance with the ethical standards and requirements of Loyola University Stritch School of Medicine.
Availability of data and material: The data that support the findings of this study are available from the corresponding author, SJ, upon reasonable request.
Author contributions: First draft of manuscript: Samantha Jakuboski; Critical review, writing and revisions: Samantha Jakuboski, Imran Uraizee, and Vijayalakshmi Ananthanarayanan.
Final review: Samantha Jakuboski, Imran Uraizee, Vijayalakshmi Ananthanarayanan, Paul Camren, Girish Venkataraman, Daniel Dilling.
Conflict of Interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Informed consent was obtained for this publication.
References
- Randall RE, Jr. Williamson WC, Mullinax F, Tung MY, Still WJ. Manifestations of systemic light chain deposition. Am J Med. 1976;60(2):293–299.
- Preud’homme JL, Aucouturier P, Touchard G, Striker L, Khamlichi AA, Rocca A, et al. Monoclonal immunoglobulin deposition disease (Randall type). Relationship with structural abnormalities of immunoglobulin chains. Kidney Int. 1994;46(4):965–972.
- Gertz MA. Immunoglobulin light chain amyloidosis: 2020 update on diagnosis, prognosis, and treatment. Am J Hematol. 2020;95(7):848–860.
- Kijner CH, Yousem SA. Systemic light chain deposition disease presenting as multiple pulmonary nodules. A case report and review of the literature. Am J Surg Pathol. 1988;12(5):405–413.
- Wei P, Tao R, Liu Y, Xie H, Jiang S, Yu D, et al. Pulmonary light chain deposition disease: a case series and literature review. Ann Transl Med. 2020;8(9):588.
- Rho L, Qiu L, Strauchen JA, Gordon RE, Teirstein AS. Pulmonary manifestations of light chain deposition disease. Respirology. 2009;14(5):767–770.
- Bhargava P, Rushin JM, Rusnock EJ, Hefter LG, Franks TJ, Sabnis SG, et al. Pulmonary light chain deposition disease: report of five cases and review of the literature. Am J Surg Pathol. 2007;31(2):267–276.
- Pantazis G, Psaras T, Krope K, von Coelln R, Fend F, Bock T, et al. Cerebral low-grade lymphoma and light chain deposition disease: exceedingly high IgG levels in the cerebrospinal fluid as a diagnostic clue. Clin Neuropathol. 2010;29(6):378–383.
- Popovic M, Tavćar R, Glavac D, Volavsek M, Pirtosek Z, Vizjak A. Light chain deposition disease restricted to the brain: The first case report. Hum Pathol. 2007;38(1):179–184.
- Stone GC, Wall BA, Oppliger IR, Wener MH, Jolly SL, Aguirre A, et al. A vasculopathy with deposition of lambda light chain crystals. Ann Intern Med. 1989;110(4):275–278.
- Stuhlinger W, Berek K, Lapin A, Jaschke E, Pastner D. [Gamma 1 heavy chain disease with immune vasculitis and rheumatoid arthritis]. Klin Wochenschr. 1987;65(8):359–368.
Keywords
Light chain deposition disease; Pulmonary cysts; Vasculitis
Cite this article
Jakuboski S, Uraizee I, Camren P, Venkataraman G, Dilling D, Ananthanarayanan V. Pulmonary Light Chain Disease: An unusual presentation in a 54-year-old man with pulmonary cysts and granulomatous inflammation with localized vasculitis. Clin Case Rep J. 2022;3(5):1–6.
Copyright
© 2022 Vijayalakshmi Ananthanarayanani. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
