Early Onset of Aggressive Primary Cutaneous CD8+ Gamma-Delta T-Cell Lymphoma at Young Female Patient with Hemophagocytic Syndrome and Fatal Pilocytic Astrocytoma Cerebellar Involvement
* Ivan Kukeev;
Elchanan Quint;
Itai Levi;
Karen Nalbandyan;
Anton Osyntsov;
Nir Cohen;
Guy Barsky;
-
* Ivan Kukeev: Department of General Surgery B, Soroka University Medical Center, Beer Sheva, Israel.
-
Elchanan Quint: Department of General Surgery B, Soroka University Medical Center, Beer Sheva, Israel.
-
Itai Levi: Department of General Surgery B, Institute of Hematology, Soroka University Medical Center, Beer Sheva, Israel.
-
Karen Nalbandyan: Department of Pathology, Soroka University Medical Center, Beer Sheva, Israel.
-
Anton Osyntsov: Department of General Surgery B, Soroka University Medical Center, Beer Sheva, Israel.
-
Nir Cohen: Department of General Surgery B, Soroka University Medical Center, Beer Sheva, Israel.
-
Guy Barsky: Department of General Surgery B, Soroka University Medical Center, Beer Sheva, Israel.
Abstract
Introduction: Primary cutaneous gamma-delta T-cell lymphoma (PCGD-TCL) is a rare lymphoma that accounts for 1% of all cutaneous T-cell lymphomas. The average age at onset of the disease is 42 years. We present a cutaneous gamma-delta T-cell lymphoma case in a 26-year-old patient with hemophagocytic syndrome and simultaneous cerebellar pilocytic astrocytoma manifestations.
Case Presentation: A 26-year-old patient has visited our emergency department several times with an abscess of the anterior abdominal wall. Biopsy of the abscess wall revealed primary cutaneous gamma-delta T-cell lymphoma. Therapy started with the CHOEP protocol. Unfortunately, two months later, the patient manifested Hemophagocytic Syndrome (HPS) and cerebellar pilocytic astrocytoma with the development of an intracerebral hemorrhage, which was fatal.
Discussion: Primary cutaneous gamma-delta T-cell lymphoma (PCGD-TCL) is an aggressive variant of cutaneous lymphoma. The simultaneous development of a central nervous system tumor and hemophagocytic syndrome are rare findings that are commonly fatal. Most patients have a dismal prognosis, even with chemotherapy and radiation. Stem cell transplantation is an increasingly promising option for these patients. The medium survival of patients is 15 months.
Conclusion: PCGD-TCL should be suspected in patients with cutaneous manifestations that persist despite conservative treatment. Physicians who deal with patients with rapidly progressive, nonspecific resistance to therapy should maintain awareness of uncommon forms of cutaneous T-cell lymphomas.
Abbreviations
PCGD-TCL: Primary Cutaneous Gamma-Delta T-cell Lymphoma; HPS: Hemophagocytic Syndrome; DIC: Disseminated Intravascular Coagulation; CT: Computed Tomography; MRI: Magnetic Resonance Imaging; WBC: White Blood Cells; HB: Hemoglobin; PLT: Platelet Count; IHC: Immunohistochemistry Test; CNS: Central Nervous System; VEGF: Vascular Endothelial Growth Factor; LDH: Lactate Dehydrogenase; FFP: Fresh Frozen Plasma; WHO-EORTC: World Health Organization-European Organization for Research and Treatment of Cancer; CTCL: Cutaneous T-cell Lymphoma; PET-CT: Positron Emission Tomography and Computed Tomography
Introduction
Primary cutaneous gamma-delta T-cell lymphoma (PCGD-TCL) is a highly aggressive tumor defined as a clonal proliferation of mature gamma-delta T-cells with cytotoxic phenotypes [1,2]. PCGD-TCL has a characteristic immunophenotype (CD3+, CD2+, CD7+/–, beta F1–), where most cells express neither CD4 nor CD8, although in isolated cases, cells may be CD8 positive. This type of lymphoma can exhibit diverse histological patterns, including epidermotropism, dermal or subcutaneous involvement with extremity preference, and aggressive clinical course [3].
The average age at the onset of the disease is 42 years [4]. Only two primary gamma delta T-cell lymphoma cases with hemophagocytic syndrome manifestation have been described [5,6]. We present a case report with the early onset of PCGD-TCL in a 26-year-old woman with the hemophagocytic syndrome and the simultaneous development of cerebellar pilocytic astrocytoma.
Case Presentation
A 26-year-old woman with a medical history of two miscarriages and overweight with complaints of weakness, periodic fever, headaches, and pain in the elbows and knees during the previous month was hospitalized in Emergency Department. An abscess of the anterior abdominal wall with a size 3 cm X 2 cm was incised. The patient was discharged to her home with a prescription for Amoxicillin/clavulanic acid for seven days. Two weeks later, she returned with complaints of fever during the last three days, swelling and redness around the incised abscess, muscle pain, and shortness of breath. Examination of the patient revealed: a fever up to 39°C and tachycardia of 112 beats per minute. An abscess appeared on the side of the abdominal wall: fluctuation, edema, and a local increase in temperature around the previous incision. Enlarged lymph nodes in the left groin area were palpable. In blood tests - hemoglobin was 11.2 g/dL, leukopenia - 3.53 x 109/L, platelets - 90 X 109/L, and an increased Lactate Dehydrogenase (LDH) level to 1383 (U/L). Computed Tomography (CT) abdomen scan revealed splenomegaly with edema of the left anterior abdominal wall with a fluid accumulation, spreading to the left groin without the involvement of the lymphatic system (Figure 1).
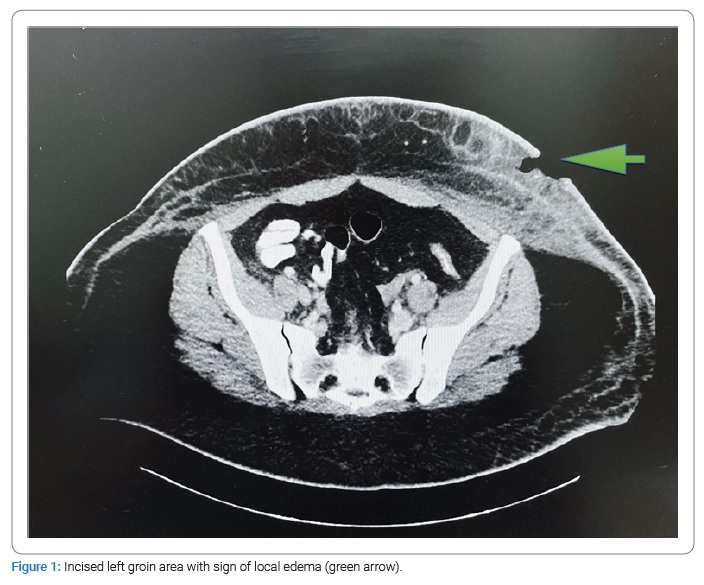
An abscess area was incised, and the clear, odorless liquid was extracted. Antibiotic therapy with Cefazolin was started without significant improvement in the clinical course. There was still no improvement after changing the antibiotics to extended-spectrum with Meropenem, Vancomycin, and Doxycycline. The surface of the wound remained with areas of necrosis and edema. Pancytopenia (WBC - 2.44 x 109/L, HB - 7.5 g/dL, PLT - 91 x 109/L) with an increase in the level of LDH to 4000 were noted in blood tests. The growth of microorganisms in the fluid abscess was not detected. Tests for HIV and brucellosis were negative. In the absence of clinical improvement, we suspected a lymphoproliferative disorder. The patient refused a bone marrow biopsy.
Due to the significant deterioration of the patient’s condition and lack of pathological diagnosis, a biopsy of the abscess wall was performed. The immunohistochemistry test (IHC) revealed CD3, CD2, CD5, CD7, CD8, TIA1, Granzyme, and TCR gamma positivity. Ki67 proliferation index was as high as 75%. Diagnosis of primary cutaneous gamma delta T-cell lymphoma was confirmed (Figure 2).
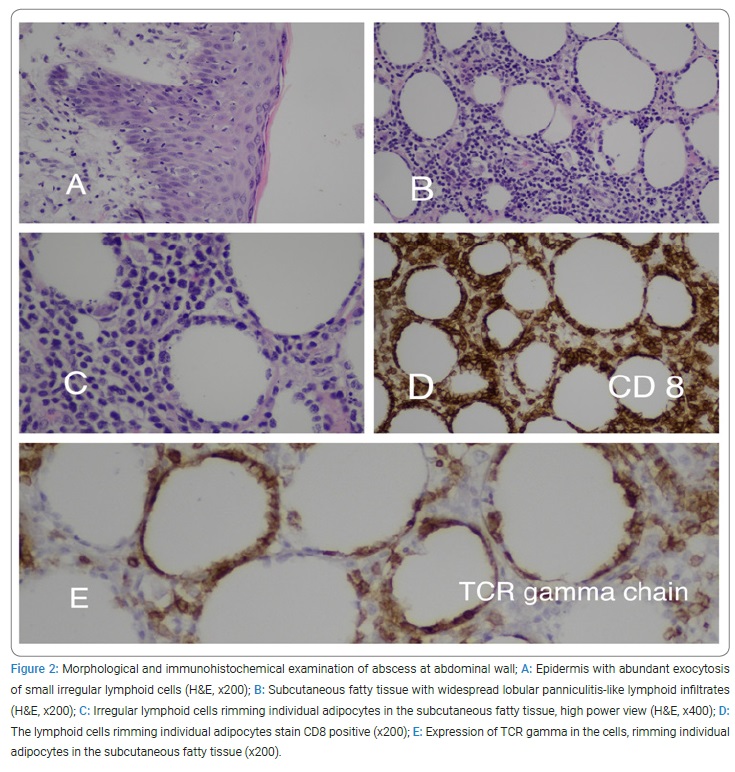
In looking for an extension of cutaneous lymphoma and involvement of the lymphatic system, Positron Emission Tomography and Computed Tomography (PET-CT) was performed, which confirmed the presence of local disease. The patient was transferred to the hematology department, and therapy was started according to the CHOEP protocol (cyclophosphamide, doxorubicin, etoposide, vincristine, and prednisone). Given the high level of ferritin - 25898 ng/ml (normal range 13 ng/ml –150 ng/ml), low-level NK activity -11% (normal range > 20%), fever, splenomegaly, and cytopenias, the patient was diagnosed with hemophagocytic lymphohistiocytosis. Dexamethasone was added to the treatment protocol. Due to the presence of Disseminated Intravascular Coagulation (DIC) and pancytopenia in the patient’s clinical course, she received blood products, including Fresh Frozen Plasma (FFP), vitamin K, and cryoprecipitate, thrombocytes, and packed cells.
In the following days, the patient complained of headaches and was later found in a stupored state. Computed Tomography (CT) and Magnetic Resonance Imaging (MRI) of the brain were performed, where appeared a mass in the left cerebellar hemisphere 33 mm X 24 mm with hemorrhage, obstructive hydrocephalus, and herniation to the tentorial notch was presented (Figure 3). The patient was transferred to the Neurosurgery Department, where she underwent cranial decompression and resection of the left cerebellar tumor. Morphological examination of removed tissue presented pilocytic astrocytoma with hemorrhage area.
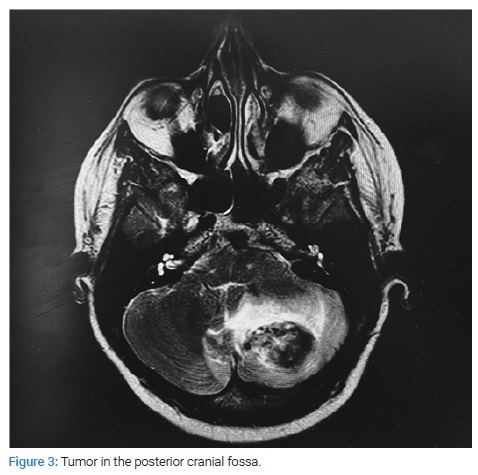
CT head examination was repeated, showing repeated bleeding from the tumor region in the posterior cranial fossa. The patient underwent an intracerebral hematoma evacuation with an external ventricular drain insertion. In the postoperative period, the patient developed acute respiratory failure and died. The death occurred two months after the primary presentation of the disease.
Discussion
Primary Cutaneous Gamma-Delta T-Cell Lymphoma (PCGD-TCL) is characterized by a clonal population of T-cells and rearrangement of TCR, which exists physiologically as a heterodimer of either alpha-beta (αβ) or gamma-delta (γδ) receptors [7–9]. PCGD-TCL accounts for less than 1% of all primary cutaneous lymphomas. It consists of an abnormal clonal proliferation of mature, activated gamma-delta T cells expressing the γδ heterodimer of the TCR. The γδ phenotype is expressed by 5% of circulating T cells and up to 30% of the total number of T cells in some organs, including the skin [10]. The updated 2017 World Health Organization (WHO) classification divided PCGD-TCLs into epidermal, dermal, and subcutaneous forms based on the location of the neoplastic infiltrate and differences in clinical courses [11].
In September 2018 was updated of the World Health Organization-European Organization for Research and Treatment of Cancer (WHO-EORTC) classification for primary cutaneous lymphomas and eight various types of Cutaneous T-cell Lymphoma (CTCL) were described, including PCGD-TCL [12]. PCGD-TCL may produce a wide range of clinical manifestations. Occurring primarily in adults, the lesions may be highly variable and range from patches and plaques to deeper subcutaneous nodules progressing to ulceration and necrosis. Extremities are preferentially involved with a relative sparing of the trunk. Systemic symptoms such as fever and night sweats are not rare findings in these patients [13,14]. This disease is typically confined to the dermis, invades deep subcutaneous areas, or may have systemic involvement (e.g., ocular, adrenal, and lymph node), but rarely invades the central nervous system (CNS) [10,15]. In the literature described, there were only five cases of cutaneous γδ T-cell lymphoma with CNS manifestations [5]. Only two patients have been described with brain involvement and died due to disease progression. Accompanying hemophagocytic syndrome was also observed in both cases. The average clinical course from the initial presentation to CNS manifestations was 17.2 months. There were no cases of simultaneous manifestation of cutaneous lymphoma with cerebellar astrocytoma.
In our case, the clinical course was characterized by a simultaneous development of intracerebellar pilocytic astrocytoma within one month from the initial appearance of PCGD-TCL. The described PCGD-TCL phenotype was highly aggressive (Ki67 - 75%). Guitar et al. showed that Granzyme B strongly correlates with a shorter survival time, and TIA-1 is associated with overall mortality [5]. In this case, both Granzyme and TIA-1 were positive.
Our patient’s disease had a rapid course which clinically was masked as an abscess of the abdominal wall. The patient died two months after the onset of the disease. Histopathologically, the disease was presented with a panniculitis-like pattern. The panniculitis-like pattern of involvement of the subcutaneous fatty tissue raised the possibility of subcutaneous panniculitis-like T-cell lymphoma. However, the cases expressing gamma-delta t-cell receptors according to the WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues should be excluded and classified as primary cutaneous gamma-delta T-cell lymphoma.
The literature describes a strong relationship between panniculitis-like PCGD-TCL and hemophagocytic syndrome [7,16]. Hemophagocytic syndrome may be congenital or associated with viral infections or malignancy. The literature mainly describes children with primary hemophagocytic lymphohistiocytosis [17]. In these children, central nervous system involvement is characterized by a histiocytic leptomeningeal and variable tissue infiltration with prominent astrogliosis [6]. The cause of our patient’s death was likely his neurological decline because of erythrophagocytosis and infiltration of the brain by gamma delta T-cells [3,16]. Considering the patient’s age, pain in the joints, the presence of miscarriages, and inhibition of three hematopoietic lineages, we do not exclude the presence of concomitant SLE in the patient, which was not diagnosed.
Our patient’s histopathological study of brain involvement discovered an intracerebellar pilocytic astrocytoma. This finding raises a logical question regarding the existence of a natural connection between PCGD-TCL and pilocytic astrocytoma. Hickman and co-authors are the only ones who have described a cerebellar pilocytic astrocytoma with the involvement of B lymphoblastic leukemia in the astrocytoma tissue. They speculate that the chemotactic migration of leukemic cells to the pilocytic astrocytoma may be partly mediated through Vascular Endothelial Growth Factor (VEGF) and VEGF receptors [18]. It is interesting to note that there is evidence of VEGF expression in peripheral T-cell lymphoma [19].
Considering the expression of TCR-γ in cutaneous T-cells, it might be associated with different clinical settings (lymphomatoid papulosis type D, mucous fungoides, and cutaneous γδ T-cell lymphoma). Therefore, it is critical to identify cutaneous T-cells that express TCR-γ or lack expression of TCR-βF1 when TCR-γ stain is unavailable [5]. The classic immunophenotype of PCGD-TCL includes expression of CD2, CD3, CD56, and TCRγ, whereas CD4, CD5, and CD6, are negative. CD8− positivity is uncommon in this entity, having been reported in only a few cases [10,20,21]. Given the CD8− positivity in our case, the differential diagnosis also included primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma.
Treatment options include a combination of radiation, immune therapy, and multiagent chemotherapy, but remission is not typically sustained with these treatment modalities [10]. Stem cell transplantation is becoming an increasingly promising option for patients with cutaneous lymphomas. A recent series shows increased survival in patients with PCGD-TCL after allogeneic and autologous stem cell transplantation [22]. Rapid diagnosis is crucial, given its aggressive clinical course with a 5-year survival rate of 20%–33% [23]. The median survival is 15 months after diagnosis [1]. It is interesting to note that a month passed from the onset of the disease to the establishment of a morphological disease in our case.
Our case highlights the importance of clinical alertness and the need for histopathologic and immunophenotypic diagnosis in case a lack of correlation of supposed disease with the clinical picture. In our patient, there was a distinctive early onset of PCGD-TCL at 26 years of age, disguised as an abscess of the abdominal wall; an aggressive course of disease (Ki67-75%); the presence of hemophagocytic syndrome, CD8− positivity with a concomitant intracerebellar manifestation of pilocytic astrocytoma, which subsequently led to intracerebral bleeding and the patient’s death two months after the onset of the underlying disease.
Conclusion
Primary Cutaneous Gamma-Delta T-Cell Lymphoma (PCGD-TCL) should be suspected in patients with nonspecific skin manifestations that persist or multiply despite conservative treatment. This disease can have a rapid, aggressive course with systemic manifestation. Despite the great rarity of this disease, the physician must have a high index of suspicion for the diagnosis of PCGD-TCL if clinical progression is inconsistent with the usual course of the supposed disease. Initial biopsies are often nondiagnostic. This type of cutaneous lymphoma often requires multiple rebiopsies with immunohistochemical analysis that became necessary to make the correct diagnosis. Further multicenter scientific studies of this pathology are required because of the high rate of progression, low survival rate, and high resistance to therapy, making PCGD-TCL one of the most insidious types of cutaneous lymphoma.
Funding
The authors received no financial support for the research, authorship, or publication of this article.
Conflict of Interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Informed consent was obtained for this publication.
References
- Koch R, Jaffe ES, Mensing C, Zeis M, Schmitz N, Sander CA. Cutaneous gamma/delta T-cell lymphoma. J Dtsch Dermatol Ges. 2009;7(12):1065–1067.
- Kempf W, Mitteldorf C. Cutaneous T-cell lymphomas-An update 2021. Hematol Oncol. 2021;39 Suppl 1:46–51.
- Toro JR, Beaty M, Sorbara L, Turner ML, White J, Kingma DW, et al. Gamma delta T-cell lymphoma of the skin: a clinical, microscopic, and molecular study. Arch Dermatol. 2000;136(8):1024–1032.
- Ralfkiaer E, Willemze R, Meijer CJLM, Dummer R, Jaffe ES. Primary Cutaneous Peripheral T-cell Lymphoma, unspecified. Pathology & Genetics of skin tumours. World Health Organization Classification of Tumours. Lyon: IARC Press. 2006. p. 184–185.
- Harrington L, Sokol L, Holdener S, Shao H, Zhang L. Cutaneous gamma-delta T-cell lymphoma with central nervous system involvement: report of a rarity with review of literature. J Cutan Pathol. 2014;41(12):936–943.
- Chakrapani A, Avery A, Warnke R. Primary cutaneous gamma delta T-cell lymphoma with brain involvement and hemophagocytic syndrome. Am J Dermatopathol. 2013;35(2):270–272.
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissue. Lyon: IARC; 2008.
- Pitch MA, Kim KH, Manning T, Smoller BR, Wong HK, Kaley JR. A diagnostically challenging case of CD8+ primary cutaneous gamma/delta T-cell lymphoma. Dermatol Online J. 2017;23(11):13030/qt0646856s.
- Gaulard P, Berti E, Willemze R, et al. Primary cutaneous peripheral T-cell lymphomas, rare subtypes. In: Swerdlow SHCE, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, et al. editors. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. p. 302–305.
- Guitart J, Weisenburger DD, Subtil A, Kim E, Wood G, Duvic M, et al. Cutaneous γδ T-cell lymphomas: a spectrum of presentations with overlap with other cytotoxic lymphomas. Am J Surg Pathol. 2012;36(11):1656–1665.
- Alberti-Violetti S, Maronese CA, Venegoni L, Merlo V, Berti E. Primary Cutaneous Gamma-Delta T-cell Lymphomas: A Case Series and Overview of the Literature. Dermatopathology (Basel). 2021;8(4):515–524.
- Willemze R, Cerroni L, Kempf W, Berti E, Facchetti F, Swerdlow SH, et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood. 2019;133(16):1703–1714.
- Foppoli M, Ferreri AJ. Gamma-delta T-cell lymphomas. European J Hematology. 2015;94:206–218.
- Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390.
- Shunmugam M, Chan E, O’Brart D, Moonim MT, Stanford MR, Morley AM. Cutaneous γδ T-cell lymphoma with bilateral ocular and adnexal involvement. Arch Ophthalmol. 2011;129:1379.
- Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105(10):3768–3785.
- Fujita Y, Sato Y, Takagi Y, Nakazato Y, Shimizu M, Mori M, et al. Hemophagocytic lymphohistiocytosis associated with primary cutaneous gamma-delta T-cell lymphoma presenting with subcutaneous panniculitis in a 12-year-old girl. Pediatr Blood Cancer. 2021;68(7):e29035.
- Hickman R, Leeman-Neill R, Rosenblum M, Anderson R, Goldman J. Co-existent pilocytic astrocytoma with acute B-cell leukemia within the cerebellum. Neuropathology. 2019;39(5):394–397.
- Foss HD, Araujo I, Demel G, Klotzbach H, Hummel M, Stein H. Expression of vascular endothelial growth factor in lymphomas and Castleman’s disease. J Pathol. 1997;183(1):44–50.
- Massone C, Chott A, Metze D, Kerl K, Citarella L, Vale E, et al. Subcutaneous, blastic natural killer (NK), NK/T-cell, and other cytotoxic lymphomas of the skin: a morphologic, immunophenotypic, and molecular study of 50 patients. Am J Surg Pathol. 2004;28(6):719–735.
- Agbay RL, Torres-Cabala CA, Patel KP, Duvic M, Quesada A, Prieto VG, et al. Immunophenotypic shifts in primary cutaneous γδ T-Cell Lymphoma suggest antigenic modulation: a study of sequential biopsy specimens. Am J Surg Pathol. 2017;41(4):431–445.
- Gibson JF, Alpdogan O, Subtil A, Girardi M, Wilson LD, Roberts K, et al. Hematopoietic stem cell transplantation for primary cutaneous γδ T-cell lymphoma and refractory subcutaneous panniculitis-like T-cell lymphoma. J Am Acad Dermatol. 2015;72(6):1010–1015.e5.
- Merrill ED, Agbay R, Miranda RN, Aung PP, Tetzlaff MT, Yuong KH, et al. Primary Cutaneous T-Cell Lymphomas Showing Gamma-Delta (γδ) Phenotype and Predominantly Epidermotropic Pattern are Clinicopathologically Distinct From Classic Primary Cutaneous γδ T-Cell Lymphomas. Am J Surg Pathol. 2017;41(2):204–215.
Keywords
Cutaneous gamma-delta T-cell lymphoma; Early onset; Hemophagocytic syndrome; CD8-positivity; Pilocytic astrocytoma
Cite this article
Kukeev I, Quint E, Levi I, Nalbandyan K, Osyntsov A, Cohen N, et al. Early onset of aggressive primary cutaneous CD8 + gamma-delta T-cell lymphoma at young female patient with hemophagocytic syndrome and fatal pilocytic astrocytoma cerebellar involvement. Clin Case Rep J. 2022;3(5):1–6.
Copyright
© 2022 Ivan Kukeev. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
