Erdheim-Chester Disease Clinically Mimicking Omental Carcinomatosis
Vishal Yalamanchili;
* Adam Bowen;
Jagmohan Sidhu;
-
Vishal Yalamanchili: Undergraduate Department of Biology, University of Rochester, Rochester, NY 14627, United States of America.
-
* Adam Bowen: Medical Student, SUNY Upstate Medical Center, NY 13210, United States of America.
-
Jagmohan Sidhu: Department of Pathology, United Health Services/Wilson Medical Center, Johnson City, NY 13790, United States of America.
Abstract
Erdheim-Chester Disease (ECD) is a rare clonal disorder of the monocyte-macrophage lineage. ECD usually presents as multifocal osteosclerotic symmetric diaphyseal and metaphyseal bone lesions with characteristic epiphyseal sparing in long bones and at least one non-osseous organ involvement. Almost any organ can be involved, but the involvement of the omentum is extremely rare. ECD presentation as an omentum-dominant disease clinically mimicking omental carcinomatosis with the sparing of metaphyses and diaphyses has not been reported in the medical literature. We describe a case of a 40-year-old male who presented with an inguinal hernia caused by herniation of a massively thickened omentum due to involvement by ECD. The diagnosis of ECD was delayed by one year because of the unusual presentation of omentum-dominant disease and lack of involvement of metaphyses and diaphyses. The patient developed Myelodysplastic Syndrome (MDS) 156 months after the diagnosis of ECD. We describe a rare presentation of ECD that caused a delay in diagnosis and discuss the relationship of ECD with MDS.
Case Presentation
A 40-year-old healthy male underwent elective right inguinal herniorrhaphy in April 2005, where thickened omentum and a nodular hernia sac were found and excised. Pathology from the hernia sac showed foci of submesothelial granulation tissue with inflammation and multinucleated giant cells. Fluid from the hernia sac showed reactive mesothelial cells with chronic inflammation and histiocytes. Slides were sent to the Armed Forces Institute of Pathology for an expert opinion which suggested histiocytic panniculitis. The differential diagnoses included pancreatitis, pancreatic carcinoma, and possible Weber Christian disease. A computerized axial tomography scan (CT scan) of the abdomen and pelvis was done in May 2005, which showed diffuse omental thickening with maximal thickness measuring 1.5 cm in the left upper quadrant, 3.3 cm in the anterior mid abdomen, 5.8 cm in the right lower quadrant and perirectal thickening. Serum amylase, lipase, carcinoembryonic antigen, and cancer antigen 19-9 were within normal ranges. Complete Blood Count (CBC) showed a White Blood Cell Count (WBC) of 7,000/uL with a normal differential, hemoglobin (hb) 14 g/dL, and platelets 354,000/uL. Mean Corpuscular Volume (MCV) was mildly low at 78 fL. Hemoglobin electrophoresis and iron profile were within normal ranges, and fecal occult blood testing was negative. Due to perirectal thickening, he underwent a sigmoidoscopy with systematic biopsies, as there was no macroscopic lesion that showed a normal sigmoid mucosa without abnormal findings or neoplasms. The patient was seen by Rheumatology. The erythrocyte sedimentation rate was 22, and an antinuclear antibody was not expressed. Complement levels were within normal ranges. The patient was started on systemic steroids for suspected Weber Christian disease.
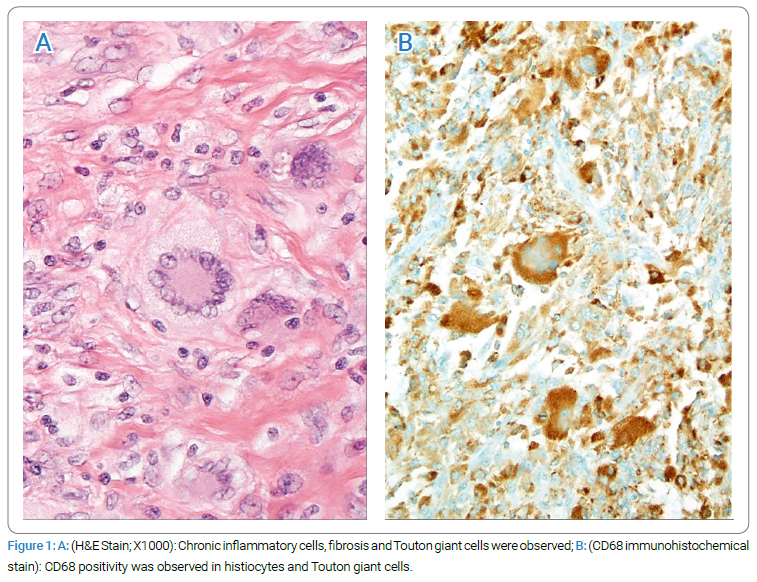
However, repeat CT scans done in February 2006 showed increased density of omental thickening, suggesting worsening disease with thickening of 1.7 cm in the left upper quadrant, 5.9 cm in the anterior mid-abdomen, and 6.2 cm in the right lower quadrant. A repeat omental biopsy again revealed panniculitis. The patient developed progressive leukocytosis and worsening anemia with each visit. In June 2006, a CBC showed a WBC of 18.9 k/uL with 81% neutrophils, hb 10.8g/dL, MCV 73 fL, and platelets 539,000/uL. A repeat iron profile showed iron 37 ug/dl, total iron binding capacity 293 ug/dl, percent saturation 12.6%, and ferritin 198 ng/mL. The bone marrow was normocellular, with findings suggestive of anemia of chronic disease and borderline megaloblastic changes in the erythroid series, suggestive of a possible drug effect. The patient developed progressive renal insufficiency with creatinine reaching up to 1.8 mg/dL in June 2006. The patient had mild edema that persisted despite stopping steroids. A 24-hour urine showed albumin of 280 mg. The renal ultrasound did not reveal any obstructive uropathy. The patient was sent to Mayo Clinic for a second opinion. They reviewed all the data from his prior evaluation and suggested a laparoscopic omental biopsy to rule out Erdheim-Chester Disease (ECD), in addition to a bone scan and bone survey. A laparoscopic biopsy done in October 2006 showed xanthogranulomatous inflammation compatible with Erdheim-Chester disease. Histiocytes, Touton giant cells, lymphocytes, and a few plasma cells diffusely infiltrated the omental tissue. Immunohistochemical stains showed that the histiocytes and Touton giant cells stained positively for CD68 and vimentin (Figure 1). Some of the lymphocytes were B-cells (CD20 and LCA positive), and most were T-cells (CD3 and LCA positive). Cytokeratin and HMB 45 were not expressed.
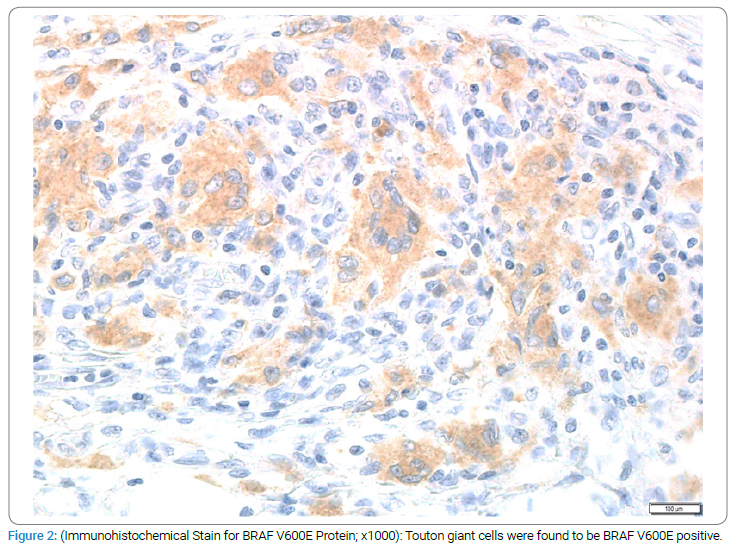
Mutational analysis of the lesional tissue from the omentum was found to be positive for BRAF V600E. The immunohistochemical stain for BRAF V600E protein was positive in the Touton giant cells (Figure 2). A bone survey showed numerous symmetrical small rounded sclerotic areas in the proximal humeri and proximal and distal femurs. A bone scan showed increased epiphyseal uptake in the knees, ankles, elbows, and shoulder joints. The patient was seen at MD Anderson in November 2006 and was started on Interferon alpha – 2A, one million units, three times a week. Repeat scans done after starting interferon showed decreasing omental thickening and less conspicuity of bone lesions. The patient’s CBC and renal function returned to normal.
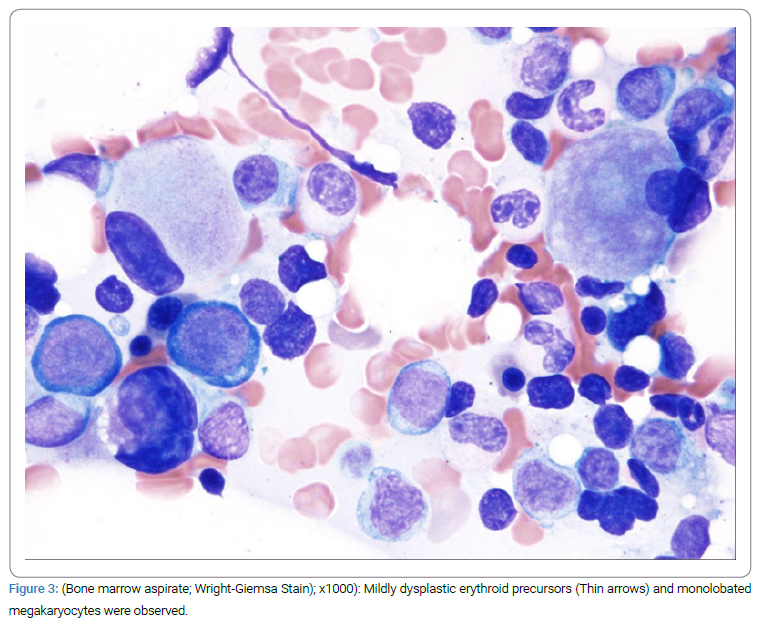
The patient continued to do well with no symptoms, and the last scans done in April 2020 showed a normal CT scan and stable bone scans and bone survey with minimal disease. However, the patient’s platelet count dropped to 123,000/uL on July 2019 and was thought to be interferon-related. The CBC in January 2020 showed a platelet count of 62,000/uL. The patient also developed lymphopenia with a lymphocyte count of 1.1 k/uL, MCV of 101, with hemoglobin declining to 12 from 14 g/dL. The bone marrow showed hypo to normocellular bone marrow with a decreased myeloid, erythroid ratio, mildly dysplastic erythroid precursors, dysplastic megakaryocytes (Figure 3), 8%–10% polytypic plasma cells with no increase in blasts. The FISH panel was positive for 5q-. The Myeloid molecular panel showed a pathogenic alteration of the TET2 gene and genomic alteration of uncertain significance in the SMC3 gene. The patient was diagnosed with 5q- MDS (myelodysplastic syndrome) 13 years (156 months) after the diagnosis of ECD.
The patient was initially treated with Lenalidomide with a good response but subsequently progressed to Refractory Anemia with Excess Blasts -1. The patient was then treated with Venetoclax with Azacytidine and received an allogenic stem cell transplant on December 20, 2022.
Resume ECD Diagnosis: The diagnosis of ECD was made, which was initially suspected to be Weber Christian disease based on the pathology from a hernia sac and thickened omentum that showed foci of submesothelial granulation tissue with inflammation and multinucleated giant cells. A CT scan of the abdomen and pelvis in May 2005 initially showed diffuse omental thickening and perirectal thickening. The patient was treated for suspected Weber Christian disease with corticosteroids. Repeat CT scans in February 2006 showed increased density of omental thickening, suggesting worsening disease. On subsequent visits, the patient developed progressive leukocytosis, worsening anemia, renal insufficiency, and some bone pain. A bone survey showed numerous sclerotic areas and increased epiphyseal uptake. A Laparoscopic biopsy of the omentum was done in October 2006, after the suggestion of Mayo Clinic, showing xanthogranulomatous inflammation and BRAF V600 E positive Touton giant cells compatible with ECD. The patient was started on Interferon alpha – 2A in November 2006, one million units three times a week, and repeat imaging showed decreasing omental thickening and less conspicuity of bone lesions. The patient’s CBC and renal function returned to normal. The patient continued to do well with no symptoms, and the last scans done in April 2020 showed a normal CT scan and stable bone scans and bone survey with minimal disease.
After the diagnosis of subsequent MDS was made, the patient’s platelet count dropped to 123,000/uL in July 2019 and then to 62,000/uL in January 2020, with lymphopenia, MCV of 101, and mildly dysplastic erythroid precursors. The patient’s bone marrow showed dysplastic megakaryocytes, polytypic plasma cells, and a positive FISH panel for 5q-. The patient was diagnosed with 5q- MDS and initially treated with Lenalidomide with good response but progressed to Refractory Anemia with Excess Blasts-1. He received treatment with Venetoclax with Azacytidine and underwent an allogenic stem cell transplant on December 20, 2022.
Discussion
ECD is considered an inflammatory myeloid neoplasm (a clonal disorder of monocyte-macrophage lineage) [1]. ECD has ERK activation in 100% of cases due to BRAF V600E mutation as a driver mutation in about one-half of cases and activating mutations in other components of MAP kinase signaling pathway as well as ECDs associated receptor tyrosine kinase (including CSF1-R mutation, NRTK1 fusion, ALK fusion, NRAS Q61R, MAP2K1, and ARAF mutation) in the other half of cases. In addition, PI3K pathway mutations also cooperate in some cases [2–4]. ECD also has a unique inflammatory cytokine signature due to the involvement of immune-mediated mechanisms that contribute to disease development and progression [5].
ECD occurs more commonly (about 75% of cases) in males and usually appears between 40 years and 70 years of age. The clinical presentation is highly heterogeneous, ranging from smoldering unifocal form to multiorgan life-threatening disease [4]. ECD usually presents as multifocal osteosclerotic symmetric long bone lesions involving only diaphyses and metaphyses (without epiphyseal involvement) and involvement of at least one non-osseous tissue, which can be almost any organ [6]. Omentum is rarely involved [4–8]. Our case’s presentation was atypical due to omental thickening and bilateral symmetric involvement of epiphyses of long bones.
An ECD Classification was proposed in 2014. It is based on clinical severity and organ system dominance. According to this classification, ECD can be divided into two major groups: asymptomatic or minimally symptomatic type and symptomatic type. Asymptomatic or minimally symptomatic types can be further divided into cutaneous-dominant disease and minimal or asymptomatic bone disease. Symptomatic ECD can be further classified based on dominant organ involvement (CNS dominant, cardiac dominant, retroperitoneal dominant, orbital-craniofacial dominant, neuroendocrine dominant, pulmonary dominant, and multisystem ECD). Omentum has rarely been described as an organ that can have involvement but has never been mentioned as a dominant organ of involvement by ECD [8].
Bone: ECD usually presents as multifocal, osteosclerotic, bilaterally symmetric, long bone lesions involving diaphyses and metaphyses (without epiphyseal involvement). Bone pain is present in half of the cases and usually manifests as mild, persistent juxta-articular pain, particularly in the lower extremities.
Cardiovascular: Cardiac involvement can be varied and includes valvular abnormalities, rhythm or conduction defects, and pericardial fibrosis. Pseudotumor of the right side of the heart and “coated aorta” due to fibrous encasement have been reported. Complications like myocardial infarction, cardiomyopathy, and symptomatic valve disease can be a major source of morbidity and mortality from ECD.
Central nervous system (CNS): Neurologic involvement is seen in up to half of cases. Infiltration can involve the entire CNS, including intra-axial and extra-axial compartments. Pyramidal and cerebellar signs are the most common neurologic symptoms. Seizures, cranial nerve palsies, headaches, neuropsychiatric symptoms, and cognitive impairment have been reported. Exophthalmos, retro-orbital pain, oculomotor palsies, or blindness can result from orbital infiltration. ECD cells can infiltrate the dura, and pituitary involvement can lead to symptoms of central diabetes insipidus and other endocrinopathies. Maxillary and sphenoid sinus involvement are also common.
Cutaneous: Skin is involved in one third of the patients. Xanthelasma and papulonodular lesions are common.
Pulmonary: Pulmonary involvement may be asymptomatic or manifest as dyspnea and/or cough. Progressive fibrosis leading to respiratory failure can occur in severe cases.
Renal: Retroperitoneal fibrosis and histiocytic infiltration in the ureters can both lead to hydronephrosis and progressive renal insufficiency. A “hairy kidney” with rind-like involvement of the perinephric tissues has been reported.
Involvement of other structures (breast, thyroid, testis, gingiva, kidneys, and spleen) is rare.
Delayed diagnosis of ECD is common due to subtle involvement of organs, lack of too many symptoms for a long time, and nondescript inflammatory-fibrotic histomorphologic features in the biopsy specimens. It can take months to decades from symptom-onset to diagnosis [4,8]. The presence of Touton giant cells admixed with chronic inflammatory cells and fibrosis helps histologically suspect ECD [8].
Progression of ECD to MDS, Acute Myeloid Leukemia (AML), or any type of Myeloproliferative Neoplasm (MPN) has been described [3,9,10]. According to Durham BH et al. the acquisition of TET2 mutation in the bone marrow stem cells or myeloid progenitors leads to clonal hematopoiesis. Secondary acquisition of the BRAF V600E mutation occurs, resulting in circulating monocytes and dendritic cells harboring BRAF V600E mutation derived from mutated myeloid progenitors. Migration of BRAF V600E-bearing dendritic cells or monocytes in tissue and differentiation into either CD1a+ langerin+ Langerhans cells (leading to Langerhans cell histiocytosis) or CD68+CD163+ foamy histiocytes (leading to ECD). The acquisition of the JAK2 V617F in the bone marrow cells/progenitors, promoted by underlying clonal hematopoiesis, leads to associated MPN/MDS. JAK2 V600E was the most common mutation identified in the study, followed by NRAS, TET2, ASXL1, and UA2F1 [3]. Moreover, recently, Ghobadi A et al. found the same BRAF V600E mutation in the bone marrow blasts of an AML patient and in ECD cells from the lung biopsy of the patient, who also had extensive involvement of organs other than lungs. Whole exome sequencing showed many shared mutations in the AML cells and the ECD cells and confirmed the origin of both AML and ECD from the same progenitor [9]. A single case of ECD with concomitant MDS showing BRAF V600E mutation in both the ECD cells and the MDS cells was identified in a recent study of 189 ECD patients with associated myeloid neoplasm in 23 patients [9]. Our case had ECD that later developed MDS with isolated 5q minus and TET2 mutation.
Treatment
A small proportion of patients may have an indolent course with no symptoms. In the absence of CNS involvement or symptoms, these patients can be observed.
Targeted therapy: In symptomatic patients, targeted therapies based on the mutational analysis is the recommended treatment of choice. For example, in patients with BRAF V600 E mutation, Vemurafenib, which is a potent inhibitor of the kinase domain of the mutant BRAF, is approved by the US Food and Drug administration [11].
In patients that have failed BRAF inhibition, MEK inhibitors have shown some promise. MEK inhibitors also hold promise for the treatment of ECD with mutations of other signaling molecules like NRAS, PIK3A, etc. Cobimetinib has been granted breakthrough designation for ECD [12].
MTOR inhibitors have shown some promise in ECD [13].
Interferon alpha: Interferon alpha is a treatment option for patients with no actionable mutations or those unresponsive to or intolerant of targeted agents [14].
Chemotherapy: Chemotherapy is usually reserved for symptomatic, fit patients refractory to the above therapies. Cladribine and low-dose methotrexate have been shown to have responses in some patients [15,16].
Glucocorticoids: Glucocorticoids have demonstrated clinical activity in ECD but have not demonstrated a survival benefit. Glucocorticoids are generally reserved for patients who cannot tolerate more aggressive systemic therapies or who have very mild symptoms.
In summary, we have described a case of a patient who had undiagnosed ECD for a year due to a rare presentation, which involved predominately the omentum and epiphysis rather than diaphyseal/metaphyseal involvement. Unfortunately, the patient developed cytopenias, which led to the diagnosis of MDS, a condition that can occur in ECD patients after the initial ECD diagnosis. Therefore, we should bear in mind that when a lesion’s histomorphology shows fibrosis and foamy histiocytes with Touton-type giant cells, we should consider ECD as a possibility. Additionally, if cytopenias develop in an ECD patient, we should consider the possibility of MDS or other myeloid malignancies.
Conflict of Interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Informed consent was obtained for this publication.
References
- Haroche J, Cohen-Aubart F, Charlotte F, Maksud P, Grenier PA, Cluzel P, et al. The histiocytosis Erdheim-Chester disease is an inflammatory myeloid neoplasm. Expert Rev Clin Immunol. 2015;11(9):1033–1042.
- Haroche J, Charlotte F, Arnaud L, von Deimling A, Hélias-Rodzewicz Z, Hervier B, et al. High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses. Blood 2012;120(13):2700–2703.
- Durham BH, Roos-Weil D, Baillou C, Cohen-Aubart F, Yoshimi A, Miyara M, et al. Functional evidence for derivation of systemic histiocytic neoplasms from hematopoietic stem/progenitor cells. Blood. 2017;130(2):176–180.
- Goyal G, Heaney ML, Collin M, Cohen-Aubart F, Vaglio A, Durham BH, et al. Erdheim-Chester disease: Consensus recommendations for evaluation, diagnosis, and treatment in the molecular era. Blood. 2020;135(22);1929–1945.
- Arnaud L, Gorochov G, Charlotte F, Lvovschi V, Parizot C, Larsen M, et al. Systemic perturbation of cytokine and chemokine networks in Erdheim-Chester disease: a single-center series of 37 patients. Blood. 2011;117(10):2783–2790.
- Munoz J, Janku F, Cohen PR, Kurzrock R. Erdheim-Chester disease: characteristics and management. Mayo Clinic Proc. 2014;89(7);985–996.
- Pan A, Doyle T, Schlup M, Lubcke R, Schultz M. Unusual manifestation of Erdheim-Chester disease. BMC Gastroenterology. 2011;11:77.
- Diamond EL, Dagna L, Hyman DM, Cavalli G, Janku F, Estrada-Veras J, et al. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood. 2014;124(4):483–492.
- Ghobadi A, Miller CA, Li T, O’Laughlin M, Lee YS, Ali M, et al. Shared cell of origin in a patient with Erdheim-Chester disease and acute myeloid leukemia. Haematologica. 2019;104(8): e373–e375.
- Papo M, Diamond EL, Cohen-Aubart F, Emile JF, Roos-Weil D, Gupta N, et al. High prevalence of myeloid neoplasms in adults with non-Langerhans cell histiocytosis. Blood. 2017;130(8):1007–1013.
- Haroche J, Cohen-Aubart F, Emile J-F, Maksud P, Drier A, Tolédano D, et al. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAF(V600E)-mutated Erdheim-Chester disease. J Clin Oncol. 2015;33(5):411–418.
- Diamond EL, Durham BH, Ulaner GA, Drill E, Buthorn J, Ki M, et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature. 2019;567(7749):521–524.
- Gianfreda D, Nicastro M, Galetti M, Alberici F, Corradi D, Becchi G, et al. Sirolimus plus prednisone for Erdheim-Chester disease: an open-label trial. Blood. 2015;126(10):1163–1171.
- Arnaud L, Hervier B, Néel A, Hamidou MA, Kahn JE, Wechsler B, et al. CNS involvement and treatment with interferon-αare independent prognostic factors in Erdheim-Chester disease: a multicenter survival analysis of 53 patients. Blood. 2011;117(10):2778–2782.
- Goyal G, Shah MV, Call TG, Litzow MR, Hogan WJ, Go RS. Clinical and radiologic responses to cladribine for the treatment of erdheim-chester disease. JAMA Oncol. 2017; 3(9):1253–1256.
- Jeon IS, Lee SS, Lee MK. Chemotherapy and interferon-alpha treatment of Erdheim-Chester disease. Pediatr Blood Cancer. 2010;55(4):745–747.
Keywords
Erdheim-Chester disease; Omentum; Carcinomatosis; Myelodysplastic syndrome
Cite this article
Yalamanchili V, Bowen A, Sidhu J. Erdheim-chester disease clinically mimicking omental carcinomatosis. Clin Case Rep J. 2023;4(2):1–6.
Copyright
© 2023 Adam Bowen. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
