Chromosome 12p12.2p11.22 Deletion in a Patient with Ventricular Fibrillation, Mitral Valve Prolapse, Dilation of Aorta and Intellectual Disability: A Case Report
Liu J;
Gajewski K;
* Upadia J;
-
Liu J: Hayward Genetics Center, Department of Pediatrics, Tulane University School of Medicine, New Orleans, Louisiana, USA; Department of Pediatrics, Tulane University School of Medicine, New Orleans, Louisiana, USA.
-
Gajewski K: Division of Pediatric Cardiology, Louisiana State University Health Sciences Center/Children’s Hospital, New Orleans, Louisiana, USA.
-
* Upadia J: Hayward Genetics Center, Department of Pediatrics, Tulane University School of Medicine, New Orleans, Louisiana, USA; Department of Pediatrics, Tulane University School of Medicine, New Orleans, Louisiana, USA.
Abstract
Interstitial deletion within chromosome 12p is a rare chromosome abnormality. Described clinical phenotypes include short stature, psychomotor delay, microcephaly, brachydactyly, and optic nerve hypoplasia. Here we report a 24-year-old female with 8050 kilobase deletion at 12p12.2p11.22, who has a history of sudden cardiac arrest secondary to cardiac arrhythmia. Cardiac features presented in our case have not been reported in 12p deletion patients. Among 40 OMIM genes in the deleted region, ABCC9 heterozygous deletion is one of the susceptible causes for the cardiac arrhythmia phenotype in this patient.
Introduction
Interstitial deletion of the proximal short arm (p) of chromosome 12 is a rare chromosome abnormality. To date, there are around 20 cases reported with chromosome 12p deletion overlapping and/or partially overlapping bands12p12p11 in the literature [1–9]. The common characteristic features reported among these patients overlap and include short stature, psychomotor delay, intellectual disability, microcephaly, digital anomalies, skeletal anomalies, dental anomalies, optic nerve hypoplasia/atrophy, strabismus, and arterial hypertension. In addition, craniofacial dysmorphic has been described, including flattened nasal bridge, epicanthal folds, cleft lip, cleft palate, and low-set ears [4,6,9]. Reported organ anomalies include cystic kidney, horseshoe kidney, and cardiac anomalies [4,5]. In addition, some of the previously described patients with proximal interstitial 12p deletion had cardiovascular abnormalities, which include high blood pressure, right heat hypoplasia, small persistent ductus arteriosus, atrial septal defect, and ventricular septal defect [3,4,9,10].
Here, we report a patient with an interstitial deletion at 12p12.2p11.22 encompassing the ABCC9 gene (loss-of-function) who had cardiac arrest secondary to cardiac arrhythmia. The presented case possibly further extends the phenotypic spectrum of proximal 12p deletion with cardiac phenotype and suggests the role of ABCC9 in cardiomyocyte electrophysiology and function.
Case Presentation
Our case is a 24-year-old Caucasian female who has a medical history of global developmental delay. She was the first child born to healthy and non-consanguineous parents. The family history and antenatal course were unremarkable. She was delivered at term. At birth, she was noted to have a cleft palate, which was repaired at age 10-month-old. The patient was globally delayed. She began to walk independently at the age of two years and speak at the age of eight years. At age 14 years, the patient was found to have a deletion of approximately 8050 kilobases in chromosome 12p12.2p11.22. She has been seen by a pediatric cardiologist since age 16 years for mitral valve prolapse, aortic root dilatation, and frequent premature ventricular contractions (PVCs). After a Holter revealed episodes of non-sustained ventricular tachycardia at age 14, she underwent hemodynamic catheterization and electrophysiology study.
Aside from her modest mitral insufficiency and aortic root dilatation, she had reassuring hemodynamics, and her Electrophysiological (EP) study revealed no inducible sustained ventricular tachycardia or fibrillation. She has been treated with atenolol 50 mg daily. At age 18 years, the patient had a cardiac arrest in her home and underwent resuscitation. She was found to be in ventricular fibrillation. She was intubated and given two defibrillator shocks, epinephrine, and amiodarone, before converting to sinus rhythm. One week later, the patient underwent Automatic Implantable Cardioverter-Defibrillator (AICD) placement. She remained on oral amiodarone therapy for some time afterward due to persistent frequent ventricular ectopy and occasional ventricular tachycardia with the inability to stop amiodarone. In February 2018, she underwent an EP study for attempted ablation of some of her more prominent foci. However, her ectopy was suppressed under anesthesia and became very infrequent, but with seven different morphologies. Therefore, no ablation could be performed at that time. During the follow-up, she was noted to have continued frequent Premature Ventricular Contractions (PVCs), Non-Sustained Ventricular Tachycardia (NSVT) (Figure 1), Mitral Valve Prolapse (MVP), and aortic root dilation. At about 23 years of age, she began to have sustained episodes of ventricular tachycardia and fibrillation that were appropriately treated with her ICD. Attempts at management included adding atenolol to her antiarrhythmic treatment and adding an atrial pacing lead to her ICD system to prevent bradycardia-related VT/VF. A genetic cardiomyopathy panel was sent, showing VUS on ABCC9. So a trial of quinidine was given, which resulted in an increase in the frequency of her ventricular tachycardia as well as profound dermatitis. She was restarted on amiodarone, and her beta blocker was changed to nadolol.
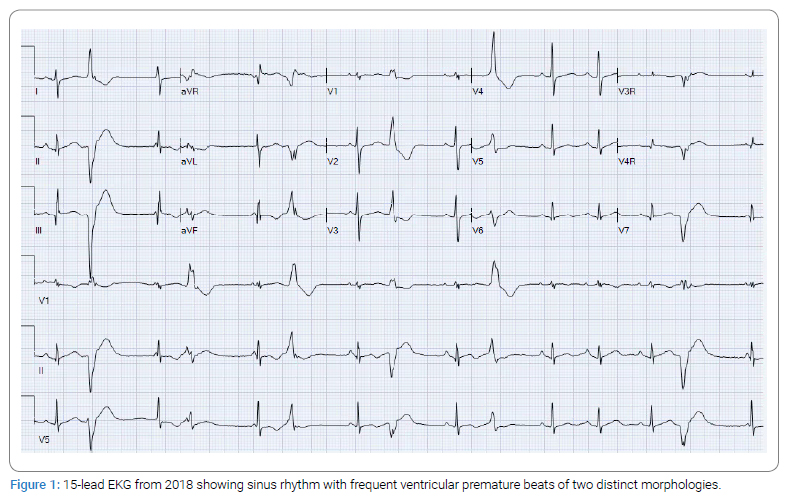
The patient has not had regression after cardiac arrest. MRI of the brain 3 months after cardiac arrest showed mild generalized cerebral atrophy. There was mild increase in size of the ventricular system and cerebral sulci when compared to previous exam prior to cardias arrest. Other medical problems include gastroesophageal reflux, gastric body and antrum hiatal hernia and conductive hearing loss, myopia, optic nerve hypoplasia, and a history of recurrent middle ear effusions as a child.
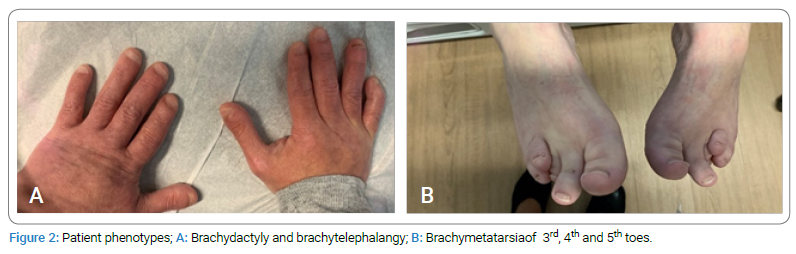
On examination at 24 years of age, her weight was 42 kilograms, height was 140 centimeters and head circumference was 51.5 centimeters. She had microcephaly, dysmorphic features including short palpebral fissures, bulbous nasal tip, flat nasal bridge, and attached earlobes. She was noted to have brachydactyly, brachytelephalanges, and brachymetatarsia of 3rd, 4th and 5th toe (Figure 2).
Results
Array Comparative Genomic Hybridization (aCGH) was performed using the Agilent 4x180K aCGH-SNP oligonucleotide array (Agilent Technologies, Santa Clara, CA), which demonstrated an approximately 8050 Kb deletion in chromosome 12p12.3p11.22 between linear genomic positions 20,834,159 and 28,884,010 (GRCh37). This deletion encompasses 47 protein-coding genes, 40 genes of which are OMIM genes.
Arrhythmia and Cardiomyopathy next-generation sequencing panel analysis using Illumina technology and deletion/duplication analysis using hybridization-based technology were performed, which include 111 genes. The result revealed a variant of uncertain significance (VUS), c.8111C>T (p.Pro2704Leu) in the ALMS1 gene; a VUS, entire coding sequence deletion in the ABCC9 gene, and a VUS, entire coding sequence deletion in KRAS gene.
Discussion
Among the cases with overlapping interstitial deletion at chromosome 12p12.2p11.2, our case represents common features of the chromosome 12p deletion, which include psychomotor delay, intellectual disability, brachydactyly, short stature, optic nerve hypoplasia, and craniofacial dysmorphism. In addition, our patient was noted to have MPV, aortic root dilation, cardiac arrhythmia, and cardiac arrest. Cardiac features seen in our case have not been reported. Our case has 8050 Kb interstitial 12p12.3p11.22 deletion, which encompasses 40 OMIM genes (gene to gene). Among these 40 genes, ABCC9, KCNJ8, and KRAS are associated with cardiovascular abnormalities.
KRAS gene (OMIM*190070, Kirsten rat sarcoma) provides instructions for making protein K-Ras which is part of the RAS/MAPK pathway. The RAS/MAPK pathway is a well-studied signal transduction pathway that plays an important role in cell growth, division, and differentiation. KRAS gene alterations have been reported as a rare cause of Noonan syndrome and cardio-facial-cutaneous syndromes. Cardiac findings observed in these cases include Atrial Septal Defect (ASD), Ventricular Septal Defect (VSD), mitral and tricuspid valve prolapse, and Hypertrophic Cardiomyopathy (HCM). Most pathogenic KRAS alterations (T58I, V14I, D153V, Y71H, and K147E) result in a gain-of-function effect and increased signal transduction down the RAS/MAPK pathway [11–15]. Loss of function deletion has not been reported in KRAS-related disorders.
KCNJ8 gene (OMIM*600935, Potassium inwardly-rectifying channel, subfamily J, member 8) encodes the Kir6.1 subunit ATP-sensitive potassium channel (KATP), allowing potassium to flow into a cell (NCBI Gene ID: 3764). Most of the publications showed evidence of gain-of-function alteration of KCNJ8 (p.S422L and p.C176S) associated with Cantú syndrome and Brugada syndrome [16–19]. Loss-of-function alterations of KCNJ8 (p.V346I and p.E332del) have been reported once as a potential mechanism in sudden infant death syndrome (SIDS) [20]. Furthermore, the deletion of chr12:21535966_21813767 (GRCh38), including KCNJ8 reported in one data entry as a polymorphism in the unaffected adult control group (DGV: nssv789559) [21].
ABCC9 gene (OMIM*601439, ATP-binding cassette, subfamily C, member 9) encodes the regulatory subunit sulfonylurea receptor 2 (SUR2) of the KATP channel. It works with the pore-forming subunit Kir6.1 to regulate the potassium channel in cardiomyocytes, vascular smooth muscles, and endothelial cells [22]. Pathogenic variants in the ABCC gene are associated with Cantú syndrome, dilated cardiomyopathy-1O, intellectual disability, and myopathy syndrome. Cantú syndrome is characterized by a coarse facial appearance, macrocephaly, hypertrichosis, and skeletal and cardiovascular anomalies [23]. The gain-of-function variants in the ABCC9 gene result in Cantu syndrome [24,25]. Additionally, gain-of-function variants (A355S, M941V, K1379Q, and H1305Y) were identified in 4 cases with sudden unexpected natural death [26].
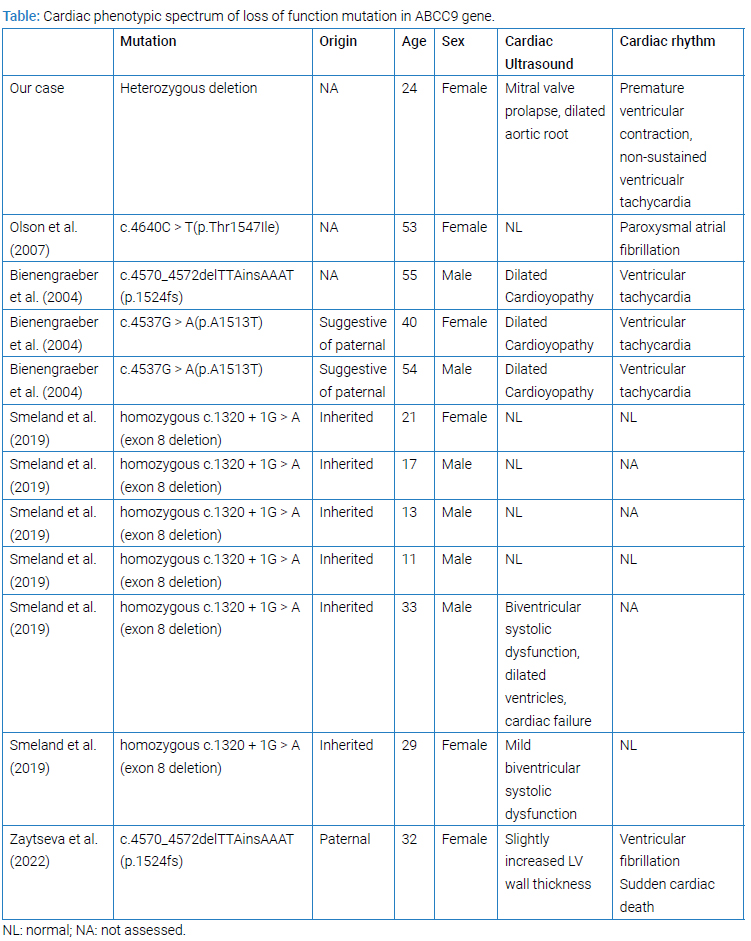
On the other hand, ABCC9 loss-of-function alternation (missense, frameshift, and deletion) has been described in 11 cases (Table). However, 6 of 11 cases were noted to have homozygous splice-site mutation in the ABCC9 gene leading to an in-frame deletion of exon 8 [27]. Only two patients out of six patients showed cardiac function abnormality, including systolic dysfunction, dilated ventricles, and cardiac failure. Cardiac conduction defects were not reported in these patients [27]. Among 5 cases with heterozygous ABCC9 alteration, three cases had missense alteration in the ABCC9 gene. Two cases had a small deletion and insertion variant resulting in frameshift alteration. In 2004, Bienengraeber identified two variants of ABCC9 in three adults, c.4570_4572delTTAinsAAAT(p.1524fs) and c.4537G>A(p.A1513T) [28]. Both variants have been approved to cause a loss of function in the ABCC9 gene. All of the three patients showed severe dilated cardiomyopathy and ventricular tachycardia. Zaytseva et al. reported the same ABCC9 variant, c.4570_4572delTTAinsAAAT(p.1524fs), in a 32-year-old female with ventricular fibrillation [22]. A missense variant, c.4640C>T(p.Thr1547Ile), was reported in a 53-year-old female who has a history of daily paroxysm of atrial fibrillation (AF) and showed an increased premature atrial ectopy in the presence of isoproterenol [29]. Even though this loss of function, ABCC9 variants in (Table) were classified as unknown significant according to the current database (ClinVar). These studies provided evidence of the relationship of ABCC9 loss-of-function alterations to cardiac phenotypes.
However, heterozygous deletion of KRAS, KCNJ8, and ABCC9 genes has not been seen in association with cardiac phenotypes. The pathologic mechanism behind our case’s cardiac phenotypes is unclear.
Conclusion
This is the first case reported to date of interstitial 12p12.2p11.22 deletion with cardiac arrhythmia and cardiac arrest. These findings possibly expand the phenotypic spectrum of interstitial 12p deletion. However, more clinical data from other patients with proximal chromosome 12p deletion are needed.
Conflict of Interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Informed consent was obtained for this publication.
References
- Orye E, Craen M. Short arm deletion of chromosome 12: report of two new cases. Humangenetik. 1975;28(4):335–342.
- Magenis E, Brown MG, Chamberlin J, Donlon T, Hepburn D, Lamvik N, et al. Resolution of breakpoints in a complex rearrangement by use of multiple staining techniques: confirmation of suspected 12p12.3 intraband by deletion dosage effect of LDHB. Am J Med Genet. 1981;9(2):95–103.
- Nagai T, Nishimura G, Kato R, Hasegawa T, Ohashi H, Fukushima Y. Del(12)(p11.21p12.2) associated with an asphyxiating thoracic dystrophy or chondroectodermal dysplasia-like syndrome. Am J Med Genet. 1995;55(1):16–18.
- Glaser B, Rossier E, Barbi G, Chiaie LD, Blank C, Vogel W, et al. Molecular cytogenetic analysis of a constitutional de novo interstitial deletion of chromosome 12p in a boy with developmental delay and congenital anomalies. Am J Med Genet A. 2003;116A(1):66–70.
- Stumm M, Klopocki E, Gasiorek-Wiens A, Knoll U, Wirjadi D, Sarioglu N, et al. Molecular cytogenetic characterisation of an interstitial deletion 12p detected by prenatal diagnosis. Prenat Diagn. 2007;27(5):475–478.
- Lu HY, Cui YX, Shi YC, Xia XY, Liang Q, Yao B, et al. A girl with distinctive features of borderline high blood pressure, short stature, characteristic brachydactyly, and 11.47 Mb deletion in 12p11.21-12p12.2 by oligonucleotide array CGH. Am J Med Genet A. 2009;149A(10):2321–2323.
- Macdonald AH, Rodriguez L, Acena I, Martinez-Fernandez ML, Sanchez-Izquierdo D, Zuazo E, et al. Subtelomeric deletion of 12p: Description of a third case and review. Am J Med Genet A. 2010;152A(6):1561–1566.
- Lee RWY, Bodurtha J, Cohen J, Fatemi A, Batista D. Deletion 12p12 involving SOX5 in two children with developmental delay and dysmorphic features. Pediatr Neurol. 2013;48(4):317–320.
- Hoppe A, Heinemeyer J, Klopocki E, Graul-Neumann LM, Spors B, Bittigau P, et al. Interstitial 12p deletion involving more than 40 genes in a patient with postnatal microcephaly, psychomotor delay, optic nerve atrophy, and facial dysmorphism. Meta Gene. 2014;2:72–82.
- Bahring S, Nagai T, Toka HR, Nitz I, Toka O, Aydin A, et al. Deletion at 12p in a Japanese child with brachydactyly overlaps the assigned locus of brachydactyly with hypertension in a Turkish family. Am J Hum Genet. 1997;60(3):732–735.
- Tidyman WE, Rauen KA. Noonan, Costello and cardio-facio-cutaneous syndromes: dysregulation of the Ras-MAPK pathway. Expert Rev Mol Med. 2008;10:e37.
- Schubbert S, Zenker M, Rowe SL, Boll S, Klein C, Bollag G, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38(3):331–336.
- Kratz CP, Zampino G, Kriek M, Kant SG, Leoni C, Pantaleoni F, et al. Craniosynostosis in patients with Noonan syndrome caused by germline KRAS mutations. Am J Med Genet A. 2009;149A(5):1036–1040.
- Niihori T, Aoki Y, Narumi Y, Neri G, Cave H, Verloes A, et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet. 2006;38(3):294–296.
- Cirstea IC, Gremer L, Dvorsky R, Zhang SC, Piekorz RP, Zenker M, et al. Diverging gain-of-function mechanisms of two novel KRAS mutations associated with Noonan and cardio-facio-cutaneous syndromes. Hum Mol Genet. 2013;22(2):262–270.
- Haissaguerre M, Chatel S, Sacher F, Weerasooriya R, Probst V, Loussouarn G, et al. Ventricular fibrillation with prominent early repolarization associated with a rare variant of KCNJ8/KATP channel. J Cardiovasc Electrophysiol. 2009;20(1):93–98.
- Medeiros-Domingo A, Tan BH, Crotti L, Tester DJ, Eckhardt L, Cuoretti A, et al. Gain-of-function mutation S422L in the KCNJ8-encoded cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave syndromes. Heart Rhythm. 2010;7(10):1466–1471.
- Cooper PE, Reutter H, Woelfle J, Engels H, Grange DK, van Haaften G, et al. Cantu syndrome resulting from activating mutation in the KCNJ8 gene. Hum Mutat. 2014;35(7):809–813.
- Veeramah KR, Karafet TM, Wolf D, Samson RA, Hammer MF. The KCNJ8-S422L variant previously associated with J-wave syndromes is found at an increased frequency in Ashkenazi Jews. Eur J Hum Genet. 2014;22(1):94–98.
- Tester DJ, Tan BH, Medeiros-Domingo A, Song C, Makielski JC, Ackerman MJ. Loss-of-function mutations in the KCNJ8-encoded Kir6.1 K(ATP) channel and sudden infant death syndrome. Circ Cardiovasc Genet. 2011;4(5):510–515.
- Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43(9):838–846.
- Zaytseva A, Tulintseva T, Fomicheva Y, Mikhailova V, Treshkur T, Kostareva A. Case Report: Loss-of-Function ABCC9 Genetic Variant Associated With Ventricular Fibrillation. Front Genet. 2022;13:718853.
- Kortum F, Niceta M, Magliozzi M, Kubat KD, Robertson SP, Moresco A, et al. Cantu syndrome versus Zimmermann-Laband syndrome: Report of nine individuals with ABCC9 variants. Eur J Med Genet. 2020;63(9):103996.
- McClenaghan C, Hanson A, Sala-Rabanal M, Roessler HI, Josifova D, Grange DK, et al. Cantu syndrome-associated SUR2 (ABCC9) mutations in distinct structural domains result in KATP channel gain-of-function by differential mechanisms. J Biol Chem. 2018;293(6):2041–2052.
- Cooper PE, Sala-Rabanal M, Lee SJ, Nichols CG. Differential mechanisms of Cantu syndrome-associated gain of function mutations in the ABCC9 (SUR2) subunit of the KATP channel. J Gen Physiol. 2015;146(6):527–540.
- Subbotina E, Yang HQ, Gando I, Williams N, Sampson BA, Tang Y, et al. Functional characterization of ABCC9 variants identified in sudden unexpected natural death. Forensic Sci Int. 2019;298:80–87.
- Smeland MF, McClenaghan C, Roessler HI, Savelberg S, Hansen GAM, Hjellnes H, et al. ABCC9-related Intellectual disability Myopathy Syndrome is a KATP channelopathy with loss-of-function mutations in ABCC9. Nat Commun. 2019;10(1):4457.
- Bienengraeber M, Olson TM, Selivanov VA, Kathmann EC, O’Cochlain F, Gao F, et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat Genet. 2004;36(4):382–387.
- Olson TM, Alekseev AE, Moreau C, Liu XK, Zingman LV, Miki T, et al. KATP channel mutation confers risk for vein of Marshall adrenergic atrial fibrillation. Nat Clin Pract Cardiovasc Med. 2007;4(2):110–116.
Keywords
Interstitial 12p deletion; ABCC9; Cardia arrhythmia; Ventricular fibrillation; Cardiac arrest
Cite this article
Liu J, Gajewski K, Upadia J. Chromosome 12p12.2p11.22 deletion in a patient with ventricular fibrillation, mitral valve prolapse, dilation of aorta and intellectual disability: a case report. Clin Case Rep J. 2023;4(3):1–5.
Copyright
© 2023 Upadia J. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
