Classic Macrophage Activation Syndrome Secondary Adult-onset Still’s Disease: Case Report
* Veghini DM;
Machado KLLL;
Lorencini PZ;
Schuwartz JP;
-
* Veghini DM: Resident, Department of Rheumatology, Cassiano Antônio de Moraes University Hospital, Brazil.
-
Machado KLLL: Assistant Physician, Department of Rheumatology, University Hospital Cassiano Antônio de Moraes, Brazil
-
Lorencini PZ: Medical student, Cassiano Antônio de Moraes University Hospital, Brazil.
-
Schuwartz JP: Medical student, Cassiano Antônio de Moraes University Hospital, Brazil.
Abstract
Adult-Onset Still’s Disease (AOSD) is a rare systemic inflammatory disorder of unknown etiology, characterized by a clinical triad of high spiking fever, arthralgia (or arthritis), and evanescent skin rash. Macrophage Activation Syndrome (MAS) is considered the most severe complication of AOSD. Management of bouth diseases poses several challenges, including difficulty in diagnosis and limited therapeutic options.
In this review, we present a case report of a patient treated with AOSD associated with MAS, with the aim of providing a better understanding of the factors involved in poor outcomes and proper care.
Introduction
AOSD is a kind of systemic inflammatory disease with unknown etiology, with a global incidence rate of (0.16–0.40)/100,000 and an estimated prevalence rate of (1-34)/1,000,000 [1,2].
The typical clinical manifestations of AOSD include Fever (60%–100%), arthritis or arthralgia (70%–100%), and maculopapular red rash (60%–80%) [2]. Other reported symptoms include myalgia, lymphadenopathy, hepatosplenomegaly, weight loss, serositis, pharyngitis, and abdominal pain [2,3,8]. Laboratory tests are characterized by elevated White Blood Cell (WBC), transaminase changes, and elevated Serum Ferritin (SF). Importantly, elevated SF is meaningful for diagnosis, especially SF increased more than 5-fold [3]. AOSD is primarily diagnosed by excluding other diseases because of the absence of markers for specific diagnosis. Yamaguchi criteria have the highest diagnostic sensitivity of 92% [2], including on major criteria [Fever ≥ 39°C persisting for ≥ 1 week, arthralgia/arthritis for ≥ 2 weeks, typical nonpruritic salmon-pink skin rash and white blood cell count ≥10×109/L (> 80% neutrophils)] and minor criteria [sore throat, lymphadenopathy and/or splenomegaly, abnormal liver function tests and negative IgM rheumatoid factor and antinuclear antibodies (immunofluorescence assay)]. For the diagnosis, ≥ 5 criteria must be met, including ≥ two major [4], requiring the exclusion of other clinical conditions that may mimic AOSD, including infection, malignancy, and other rheumatologic disorders such as autoimmune diseases and systemic vasculitis [4–6].
MAS is one of the most common complications of AOSD, with an incidence rate of over 10% [2]. It is characterized by a cytokine storm, haemophagocytosis, and multi-organ damage [3]. Its pathogenesis involves the activation of key innate immune pathways, including IL-1, IL-6, and IL-18, leading to systemic inflammation [3]. MAS is a group of clinical manifestations with high Fever, lymphadenopathy, hepatosplenomegaly, decreased blood cells, abnormal liver function, elevated serum ferritin (SF), and high triglycerides (TGs) [3,7]. MAS can increase the mortality of AOSD. Previous studies have shown that the mortality of patients with AOSD-MAS and the mortality of patients with AOSD is 52.9% and 9.5%, respectively [3,7].
Given the severity of MAS, early diagnosis and adequate management are essential in cases of AOSD. This review discusses a case report of a patient diagnosed with AOSD associated with MAS, with the aim of promoting a better understanding of the factors involved in poor prognosis and proper care.
Case Presentation
A 48-years-old woman was hospitalized with fever ≥ 39°C that started one month earlier associated with pruritic confluent macropapular rash on upper limbs, trunk and back and polyarthritis on hands, knees and ankles and asymmetrical polyarthralgia of small and medium-sized joints starting 2 weeks ago. Serologies were performed for viral hepatitis, HIV, syphilis, arboviruses, aspergillosis, histoplasmosis, and paracoccidioidomycosis, and all results were negative. Other infections, including endocarditis and negative cultures (blood and urine cultures), were excluded. Laboratory results show leukocytosis (26.740 μL) with > 80% neutrophils, hyperferritinemia (17.150 μL), increased liver enzymes, and negative rheumatoid factor and antinuclear factor. The clinical and laboratory findings confirmed the diagnosis of Adult-onset Still’s Disease.
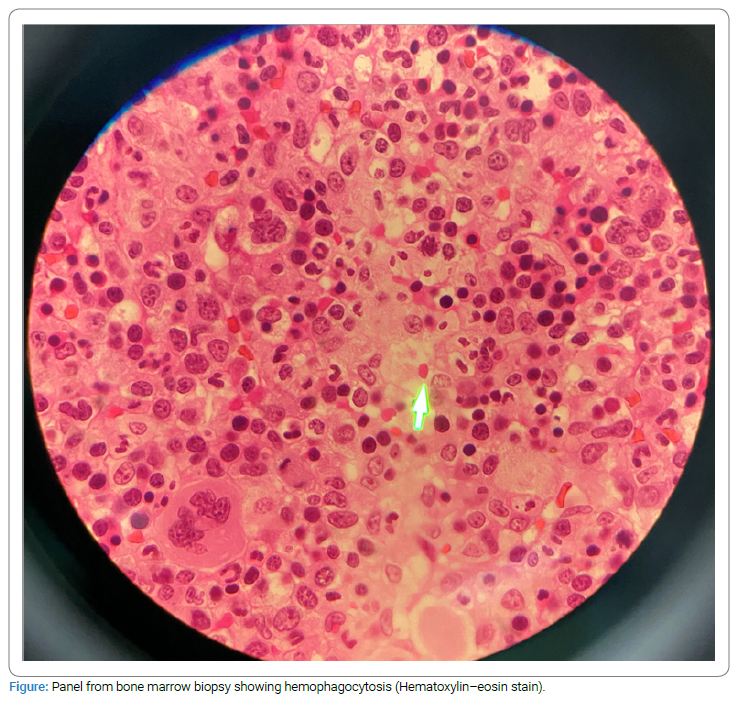
The patient was treated with 1 mg/kg of oral prednisone to attenuate the systemic condition. Later evolved with pancytopenia and daily fever, suggestive of MAS, and was confirmed with a red bone marrow biopsy which showed granulocytic hyperplasia and hemophagocytic syndrome (Figure). Furthermore, the immunophenotypic study identified T lymphocytosis at the expense of CD8 T lymphocytes, without phenotypic and polyclonal aberrations to TRBC1, in addition to the absence of CD25, which may correspond to a condition secondary to an inflammatory/infectious process. After ten days, she returned with daily fever and worsening laboratory findings, which improved after pulse therapy of methylprednisolone 1 g/day for three days.
Discussion
AOSD is a rare systemic inflammatory condition that is poorly recognized. Significant advances have been made in understanding the disease’s pathogenesis, but important unmet needs related to diagnosis and optimal management remain [5]. There is a paucity of robust epidemiological data on AOSD, and the true disease burden is difficult to estimate owing to the lack of disease registries and prospective databases and considering that most of the published data are retrospective in nature [5].
Chemokines and proinflammatory cytokines, including interferon (IFN)-γ, tumor necrosis factor α (TNFα), interleukin (IL)-1, IL-6, IL-8, and IL-18, play a crucial role in the progression of illness, resulting in the development of innovative targeted therapeutics [6].
AOSD poses many diagnostic challenges as it presents a combination of nonspecific symptoms that a wide variety of other diseases can cause. The lack of pathognomonic serologic or clinical disease markers exacerbates diagnostic delays and disease complications [5].
Patients with AOSD experience life-threatening complications, which may rapidly evolve into multiple-organ failure and death. These patients frequently developed MAS, a secondary form of Hemophagocytic Lymphohistiocytosis (HLH). The latter is characterized by continuous high fever, extreme hyperferritinemia, pancytopenia, and histopathological evidence of hemophagocytosis by activated macrophages, typically in the bone marrow [8].
The characteristics of MAS are very similar to those of AOSD, so it is very difficult to diagnose AOSD with MAS early. Because of its nonspecific clinical characteristics, MAS is not easily distinguished from other diseases, such as systemic infection and malignancy [3].
There are no treatment guidelines for AOSD due to its rarity, absence of controlled research, and lack of a standard definition for remission and therapy objectives. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs), Corticosteroids (CS), and Conventional Synthetic Disease-Modifying Antirheumatic Drugs (csDMARDs) are used in AOSD treatment. Biological therapy, including IL-1, IL-6, IL-18, and IL-17 inhibitors, as well as TNFα or Janus-Kinases (JAKs) inhibitors, is administered to patients who do not react to CS and csDMARDs or achieve an inadequate response [6,9].
In general, the prognosis of AOSD is favorable, but there is still a mortality rate of 3%, and some serious complications can also lead to death, such as MAS (12%–14%) [2,6].
Conclusion
Early recognition of Macrophage Activation Syndrome (MAS) in Adult-onset Still’s Disease (AOSD) patients still remains diagnostically challenging as there is no diagnostic test or even a set of uniform disease diagnostic criteria to differentiate MAS from the underlying systemic inflammatory condition. Although the understanding of AOSD has evolved over the last decade, there are still significant gaps in our understanding of its diagnosis, most useful biomarkers, and treatment approach. Future research in this disease area should aim for the identification and validation of tools for the early diagnosis of AOSD and optimized treatment to prevent chronic articular inflammation as well as irreversible joint damage.
Multiple recent lines of evidence have suggested new insights into AOSD pathogenesis with new therapeutic targets highlighted, thus possibly improving AOSD management in the next future.
AOSD is a rare disease that is challenging to treat but even more difficult to diagnose. This report highlights a case of AOSD associated with MAS that was refractory to oral corticotherapy. The successful use of pulse therapy emphasizes the importance of timely and appropriate management of AOSD and MAS, which can present as a cause of fever of obscure origin and can have fatal consequences if not properly treated.
Limitations
Overall, there were some limitations in this case report concerning the quality of the data found in the literature. The majority of the articles were case reports, case series, and cohort or case-control studies. In addition, heterogeneity was evident among the articles owing to a lack of standard assessment parameters.
Ethical Approval
This case report was carried out following the recommendations of the ethics committee of the Cassiano Antonio Moraes University Hospital - HUCAM, Brazil.
Consent
Written informed consent was obtained from the patient for the publication of this case report.
Conflict of Interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Informed consent was obtained for this publication.
References
- McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoS Med. 2006;3(8):e297.
- Qin A, Sun J, Gao C, Li C. Bibliometrics analysis on the research status and trends of adult-onset Still’s disease: 1921-2021. Front Immunol. 2022;13:950641.
- Gao Q, Yuan Y, Wang Y, Jiang J, Ye Z, Liu T, et al. Clinical characteristics of macrophage activation syndrome in adult-onset Still’s disease. Clin Exp Rheumatol. 2021;39 Suppl 132(5):59–66.
- Giacomelli R, Ruscitti P, Shoenfeld Y. A comprehensive review on adult onset Still’s disease. J Autoimmun. 2018;93:24–36.
- Efthimiou P, Kontzias A, Hur P, Rodha K, Ramakrishna GS, Nakasato P. Adult-onset Still’s disease in focus: Clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021;51(4):858–874.
- Macovei LA, Burlui A, Bratoiu I, Rezus C, Cardoneanu A, Richter P. Adult-onset still’s disease-a complex disease, a challenging treatment. Int J Mol Sci. 2022;3(21):12810.
- Bojan A, Parvu A, Zsoldos IA, Torok T, Farcas AD. Macrophage activation syndrome: A diagnostic challenge (Review). Exp Ther Med. 2021;22(2):904.
- Di Cola I, Ruscitti P, Giacomelli R, Cipriani P. The pathogenic role of interferons in the hyperinflammatory response on adult-onset still’s disease and macrophage activation syndrome: paving the way towards new therapeutic targets. J Clin Med. 2021;10(6):1164.
- Peckham D, Scambler T, Savic S, McDermott MF. The burgeoning field of innate immune-mediated disease and autoinflammation. J Pathol. 2017;241(2):123–139.
Keywords
Still’s disease; Adult-onset; Macrophage activation syndrome; Hemophagocytic lymphohistiocytosis; Hyperferritinemia; Autoinflammatory disorder
Cite this article
Veghini DM, Machado KLLL, Lorencini PZ, Schuwartz JP. Classic macrophage activation syndrome secondary adult-onset Still’s disease: case report. Clin Case Rep J. 2023;4(3):1–4.
Copyright
© 2023 Veghini DM. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
