RANBP2 Mutation and Acute Necrotizing Encephalopathy: Case Series
* Alqurayyan RA;
* Alamoudi RH;
Aldraihem AI;
Zakri AK;
-
* Alqurayyan RA: Department of Radiology, King Fahad Medical City, Altakhassusi Alliance Medical, Riyadh, Saudi Arabia.
-
* Alamoudi RH: Department of Radiology, King Fahad Medical City, Altakhassusi Alliance Medical, Riyadh, Saudi Arabia.
-
Aldraihem AI: Department of Radiology, King Fahad Medical City, Altakhassusi Alliance Medical, Riyadh, Saudi Arabia.
-
Zakri AK: Department of Radiology, King Fahad Medical City, Altakhassusi Alliance Medical, Riyadh, Saudi Arabia.
Abstract
Acute Necrotizing Encephalitis (ANE) is a rare but unique clinical presentation that is associated with morbidity and mortality. A virus-associated febrile illness commonly occurs prior to the onset of ANE, which is then followed by a fast decline in condition. The majority of ANE-reported cases are sporadic. However, there is still a lack of studies focusing on rare familial cases. A strong effort was undertaken to increase public awareness of this condition, as early detection and effective patient care largely depend on significant clinical judgment.
Abbreviations
ANE: Acute Necrotizing Encephalopathy; ADEM: Acute Disseminated Encephalomyelitis; OD: Autosomal Dominant; MOG: Myelin Oligodendrocyte Glycoprotein Anti-Bodies; PICU: Pediatric Intensive Care Unit; LP: Lumbar Puncture; IVIG: Intravenous Immunoglobulin
Introduction
Acute Necrotizing Encephalopathy (ANE) is a rare but distinctive type of medical condition, first proposed by Mizguchi et al. in 1995 [1]. It is generally considered a parainfectious disease as the disease appears preceding a viral infection, including parainfluenza, influenza A/B, human herpesvirus, varicella, and enterovirus, but direct involvement of the central nervous system by these agents is rare [2,3]. It also rarely occurs in bacterial infections, including Mycoplasma pneumoniae, tetanus, and pneumoniae [4]. For children, it is a rapidly deteriorating encephalopathy that is observed after acute infections between 6 years and 18 years of age. The disease presents with early-onset seizures, fever, focal neurological deficits, personality changes, and rapid deterioration of consciousness, which may ultimately progress into a coma. The majority of the first reported ANE cases were in children from Japan and Taiwan, raising the possibility that racial characteristics were involved in the condition. However, more cases—including adult cases—were subsequently documented in Western nations, demonstrating that ANE has spread globally and has no racial preference. ANE has not been recognized primarily as a clinicopathologic disorder with unknown etiology [3,5]. In line with this, recent studies have demonstrated that both genetic and environmental factors play a role in acquiring ANE.
A pathogenic variation in the RANBP2 gene causes infection-induced acute encephalopathy 3 (IIAE3, MIM #608033), an autosomal dominant condition with decreased penetrance. ANE1, which often manifests as bilateral symmetric thalamic, midbrain, and hindbrain lesions and typically occurs between 1 day–4 days post-acute viral infection, is more likely to occur before the age of 6 in those with IIAE3 [6]. The majority of cases go through distinct prodromal, acute, and recovery phases. At the prodromal stage, a non-specific febrile viral-like sickness that lasts up to 5 days is frequently observed. Following the first infection, there is an acute phase that includes seizures, localized neurological dysfunction, worsening consciousness, and coma. The ultimate stage of recovery can include complete resolution of the brain lesions, atrophy, hemosiderin deposition, or white matter cyst formation. Most cases of ANEC are nonrecurrent and sporadic [6,7]. However, the RAN-binding protein 2 (RANBP2)-associated type (ANEC1) is the rarer, genetic, and familial type. In a study by Neilson et al. 2003, 11 individuals from the same family were diagnosed with ANEC. It was stated that this disease could be inherited as autosomal dominant. Among ten unrelated individuals with ANEC who were diagnosed in 2009, the RANBP2 gene mutation was discovered for the first time. All tissues generate RANBP2, a nuclear protein with a wide range of intracellular functions. The autosomal dominant (OD) gene ANEC1 has incomplete penetrance. As a result, a RANBP2 mutation increases a person’s lifetime risk of developing ANEC1 by 40%.
It should be noted that these patients have relatives who were followed up with diagnoses such as Leigh’s disease, acute disseminated encephalomyelitis (ADEM), encephalitis, or aseptic meningitis. The RANBP2 gene has several functions, including those related to mitochondrial, metabolic, and nuclear signals, and based on the underlying kind of mutation, its alterations result in various clinical symptoms [6–8]. Blood-brain barrier dysfunction may be observed if the nuclear signal function is disrupted by mutation. A RANBP2 mutation that results in mitochondrial malfunction can also result in cell death due to a lack of energy. As a result, the disease’s involvement pattern mimics that of disorders that deplete energy, such as Wernicke’s encephalopathy and Leigh syndrome [9]. Considering the rarity of ANE as well as the lack of sufficient data on familial cases, we gathered data on one patient case of familial ANE associated with RANBP2 gene mutation.
Case Presentation
This is a case of a 3-year-old patient, full-term, delivered via Caesarian Section (C/S) with normal development. The patient was free of symptoms two weeks ago but experienced a high fever, sore throat, and ear pain three days before admission. The patient then presented with vomiting and bilateral leg pain on the day prior to admission. On the third day, the patient was unable to move his upper limbs and was not able to sit, stand, or walk (+) for squint. No reports of dysphagia, facial weakness, a history of falls or loss of consciousness, or abnormal movement or seizures were reported. He was then referred to the King Fahad Medical City, Riyadh, where he was provided with the necessary examinations. Further workup, CSF analysis, genetic testing, and follow-up MRI were done.
Upon examination, the patient presented as encephalopathic, irritable, non-cooperative, and with a decreased level of consciousness (Glasgow Coma Scale = 11). On physical assessment, the patient showed no abnormalities except that he had increased muscle tone in his left upper limbs and bilateral legs after stimulation. For laboratory tests, Culture, Myelin Oligodendrocyte Glycoprotein Anti-Bodies (MOG), Anti-NMO, and Aquaporin 4 Anti-Bodies were negative, and glucose and protein tests were normal. CSF Analysis and WBC revealed no count due to clotting (Figure 1A,Figure 1B,Figure 1C).
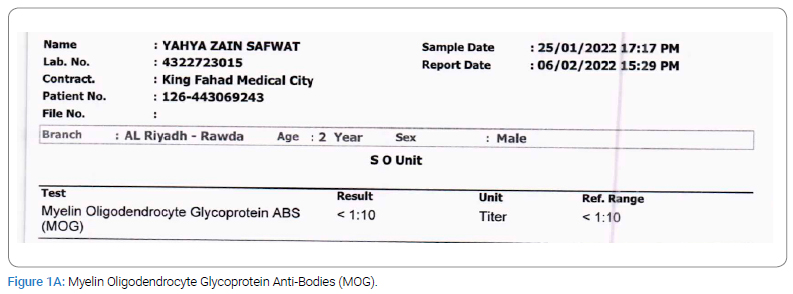
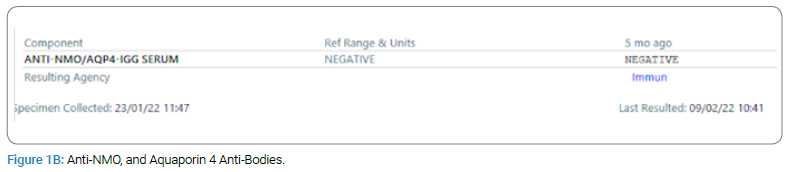
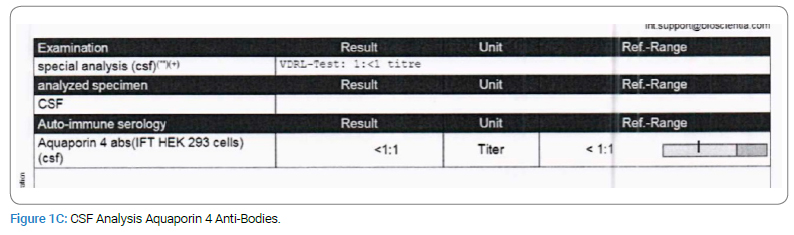
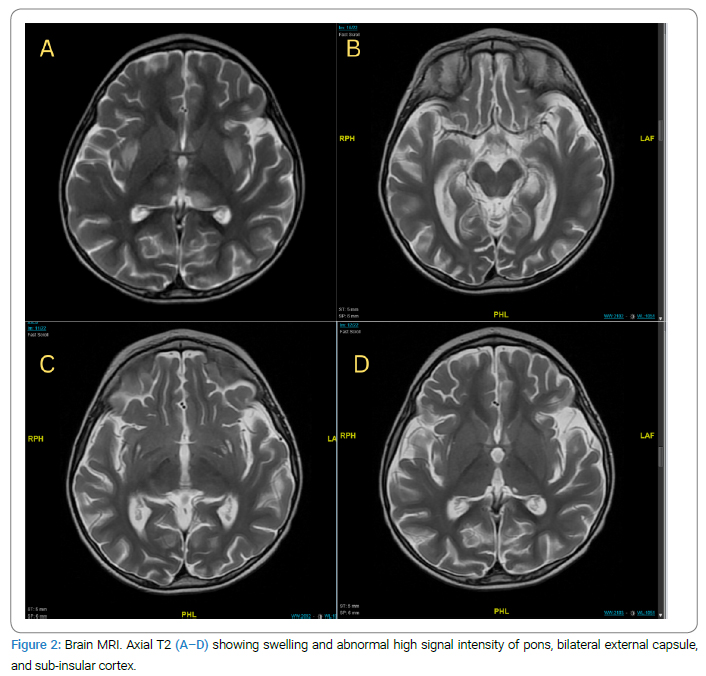
The parents denied a history of autoimmune disease in the family; however, detailed a pertinent family history where the patient’s older brother, 11-year-old old has a history of febrile illness (H1N1) followed by aphasia and weakness with similar presentations when he was 5-year-old; admitted to PICU and required ventilation, LP was negative, MRI showed Necrotizing Encephalitis (Figure 2A,Figure 2B,Figure 2C,Figure 2D).
In addition, a genetic workup and Whole Exome Sequencing (WES) were done and showed: a familial pathogenic variant in the RANPBP2 gene in the heterozygous state. The genetic diagnosis of autosomal dominant susceptibility to acute, infection-induced encephalopathy type 3 is confirmed (Figure 3).

The patient received IVIG, pulse steroid, and antibiotics with clinical improvement of motor function, and a tapering dose of prednisolone.
Discussion
Acute Necrotizing Encephalopathy of Childhood (ANEC) is a medical condition commonly seen in young, previously healthy children. Our case was a previously healthy child whose condition initiated with high fever and fast neurologic disturbances [10].
The clinical course of ANE can be categorized as the prodromal stage, acute encephalopathy, and recovery stage. ANE in its prodromal stage is diverse due to and non-specific to viral infections, including fever, vomiting, diarrhea, upper respiratory tract infection, chest infection, and headache. With progressing ANE, seizure, focal neurologic deficits, and disturbance of consciousness may present as brain dysfunctions. The clinical course of ANE is also diverse, from a mild form of encephalopathy to a debilitating form leading to death [2]. The RANBP2 mutation is autosomal dominantly inherited, yet incomplete penetrance with a 40% rate has been noted. Even among family members with the same RANBP2 gene mutations, including a set of identical twins [6], the onset, presentation, and features of the disease can differ. A number of theories have explained the incomplete penetrance. According to Neilson et al. dietary status, viral infection routes, and various viral inoculums can all affect an individual’s susceptibility to illness presentation. They postulated that changes in temperature sensitivity and altered protein function could occur from amino acid substitutions brought on by RANBP2 mutation. As a result, the length and severity of the fever may be a factor in inadequate penetration. As described earlier, clinical manifestations vary in the family. The patient is part of twins, but his other twin is reported to be healthy. One relative showed a pathogenic variant in the RANPB2 gene, and an older brother had similar presentations. However, the parents did not have clinically apparent neurologic symptoms. Due to the complexity and rarity of the medical disease, the pathophysiology is poorly known. However, the predominant theory holds that a trigger in genetically vulnerable populations with RANBP2 mutations results in abnormal nuclear signaling, which in turn causes a cytokine storm, which allows cytokines into the central nervous system, disrupts the blood-brain barrier, and results in encephalopathy [6,11]. The triggering event is most commonly viral, such as influenza A and B, parainfluenza, varicella, herpes simplex virus, enterovirus, rotavirus, rubella, and measles [2].
Critical care management is necessary for the treatment of ANE. Immune-mediated CNS cell damage in ANE has been reduced with the administration of methylprednisolone, IVIG, and other immunosuppressants. According to earlier studies, children with ANE had better outcomes when corticosteroids were administered within 24 hours after the onset of symptoms. There are few experiences with therapeutic interventions and patient follow-up for those with ANEC1, and these experiences are often case series. An improved prognosis may be associated with early steroid therapy, most likely as a result of decreasing increased cytokines and reducing inflammation. High-dose steroids can also reduce neuroinflammation-related edema. A complete recovery, chronic sequelae, or death can be the result of ANEC1. 30% of ANEC1 suffer mortality [5,12].
Key Results
In this case study, a 3-year-old patient presented with several triggers of Acute Necrotizing Encephalopathy (ANE) that have responded well to IVIG, pulse steroids, and antibiotics. MRI revealed necrotizing encephalitis, while genetic workup (whole exome sequencing) showed a familial pathogenic variant in the RANBP2 gene in the heterozygous state. No reports of dysphagia, facial weakness, a history of falls or loss of consciousness, or abnormal movement or seizures were reported.
Summary Statement
The RANBP2 gene can be considered a major cause of familial and recurrent ANE; therefore, RABNP2 mutation testing should be highly explored to improve clinical outcomes and prognosis further.
Conflict of Interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Informed consent was obtained for this publication.
References
- Mizuguchi M, Abe J, Mikkaichi K, Noma S, Yoshida K, Yamanaka T, et al. Acute necrotising encephalopathy of childhood: a new syndrome presenting with multifocal, symmetric brain lesions. J Neurol Neurosurg Psychiatry. 1995;58(5):555–561.
- Paktinat M, Hessami K, Inaloo S, Nemati H, Katibeh P, Nejabat M, et al. Case Report of RANBP2 mutation and familial acute necrotizing encephalopathy. Int J Pediatr. 2021;2021:6695119.
- Wu X, Wu W, Pan W, Wu L, Liu K, Zhang HL. Acute necrotizing encephalopathy: an underrecognized clinicoradiologic disorder. Mediators Inflamm. 2015;2015:792578.
- Sarigecili E, Ucar HK, Havali C, Cansu A, Aydin K. Acute necrotizing encephalopathy associated with RANBP2 mutation: value of MRI findings for diagnosis and intervention. Acta Neurol Belg. 2023;123(2):571–582.
- Aksoy E, Öztoprak Ü, Çelik H, Özdemir FMA, Özkan M, Kayılıoğlu H, et al. Acute necrotizing encephalopathy of childhood: a single-center experience. Turk J Med Sci. 2021;51(2):706–715.
- Hartley M, Sinha A, Kumar A, Aliu E, Mainali G, Paudel S. Acute necrotizing encephalopathy: 2 case reports on RANBP2 mutation. Child Neurol Open. 2021;8:2329048x211030751.
- Neilson DE, Adams MD, Orr CM, Schelling DK, Eiben RM, Kerr DS, et al. Infection-triggered familial or recurrent cases of acute necrotizing encephalopathy caused by mutations in a component of the nuclear pore, RANBP2. Am J Hum Genet. 2009;84(1):44–51.
- Levine JM, Ahsan N, Ho E, Santoro JD. Genetic acute necrotizing encephalopathy associated with RANBP2: clinical and therapeutic implications in pediatrics. Mult Scler Relat Disord. 2020;43:102194
- Singh RR, Sedani S, Lim M, Wassmer E, Absoud M. RANBP2 mutation and acute necrotizing encephalopathy: 2 cases and a literature review of the expanding clinico-radiological phenotype. Eur J Paediatr Neurol. 2015;19(2):106–113.
- Salehiomran MR, Nooreddini H, Baghdadi F. Acute necrotizing encephalopathy of childhood; a case report. Iran J Child Neurol. 2013;7(2):51-54.
- Shukla P, Mandalla A, Elrick MJ, Venkatesan A. Clinical manifestations and pathogenesis of acute necrotizing encephalopathy: the interface between systemic infection and neurologic injury. Front Neurol. 2021;12:628811
- Yuan P, Zhong M. Acute necrotizing encephalopathy in children: A case report and literature review. Int J Pediatr Res. 2018;4:45.
Keywords
Necrotizing encephalitis; Genetics; Pediatrics; RANBP2 mutation; Neuroimaging
Cite this article
Alqurayyan RA, Alamoudi RH, Aldraihem AI, Zakri AK. RANBP2 mutation and acute necrotizing encephalopathy: Case series. Clin Case Rep J. 2024;5(1):1–5.
Copyright
© 2024 Alqurayyan RA. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
