Abstract
Background: Alkaptonuria is an inherited metabolic disease with consequent accumulation of homogentisic acid in tissues.
Aim: Exuberant clinical presentation.
Case Presentation: A 54-year-old female presents with bluish pigmentation in her ears, fingers, and nail bed. Skin anatomopathological examination and evaluation of organic acids in urine are compatible with alkaptonuria.
Discussion: Alkaptonuria manifests with characteristic skin changes. We illustrate a case with an exuberant clinical presentation with dermatological and otological manifestations.
Introduction
Alkaptonuria, also known as endogenous ochronosis, is an extremely rare autosomal recessive metabolic disease [1,2].
Signs and symptoms usually appear in the third or fourth decades of life, with ochronotic osteoarthropathy being the most commonly reported manifestation [1,2]. The cutaneous presentation is considered a marker of the disease and should alert to the involvement of other organs [3].
Case Presentation
Female, 54-year-old, consanguineous parents, reports the appearance of bluish pigmentation in the ears, on the side of the second finger of both hands, and the nail 20 years ago—mother and sister with similar pigment changes.
The patient, who previously had osteoarthritis and osteoporosis, presented bilateral hearing loss under investigation and a history of cholecystectomy due to cholelithiasis.
She denied any other systemic alterations and used only vitamin D, calcium, and bisphosphonates, ruling out any possible exogenous exposure associated with the appearance of skin alterations.
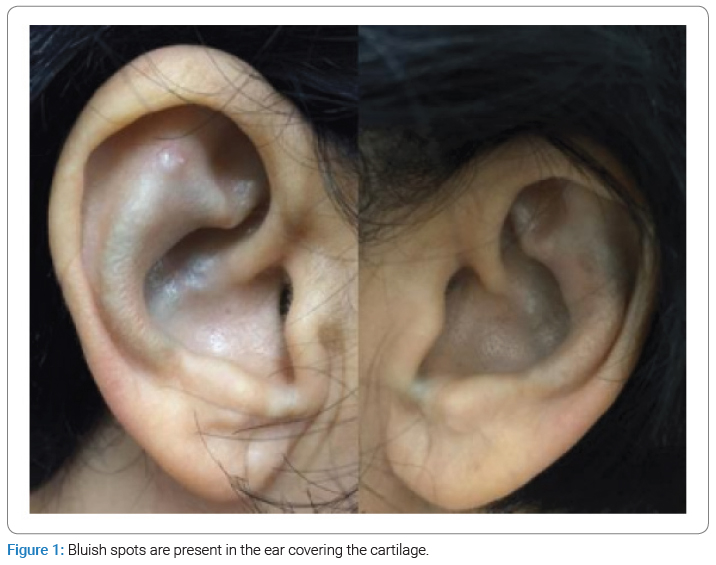
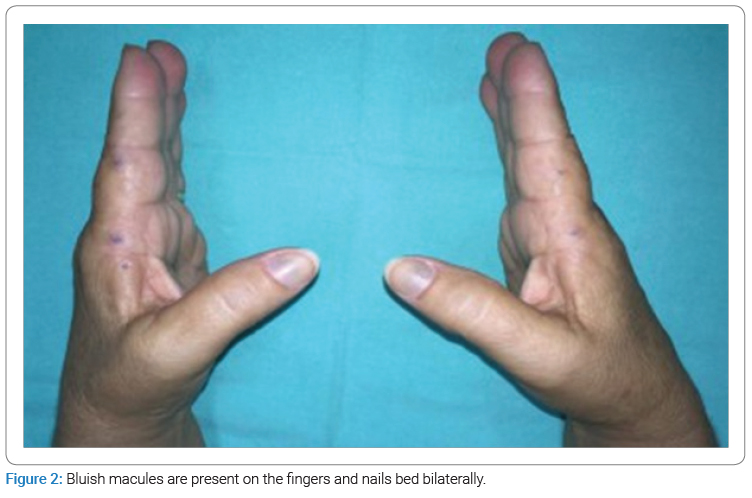
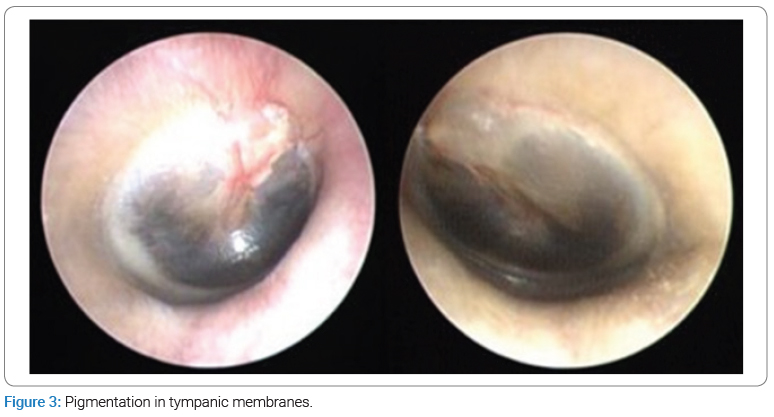
The dermatological examination showed bluish patches in the ear pavilions bilaterally (Figure 1) and bluish macules of varying sizes in fingers and nails bilaterally (Figure 2). In the otoscopic evaluation, the presence of pigmentation was detected on both tympanic membranes (Figure 3), and the audiological opinion was of moderate bilateral sensorineural hearing loss.
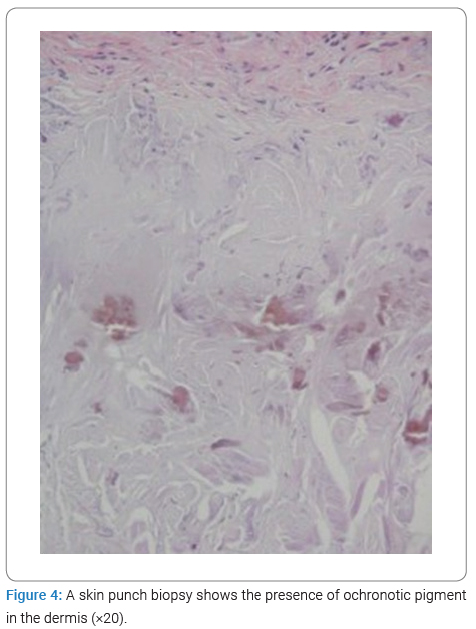
A skin biopsy was performed, which showed ochronosis in the anatomopathological examination (Figure 4). The evaluation of organic acids in the urine showed a marked increase in homogentisic acid, defining the diagnosis.
Discussion
Alkaptonuria results from a deficiency of the homogentisate enzyme 1,2- dioxygenase with the consequent accumulation of Homogentisic Acid (HA) [3]. The oxidation of HA leads to polymerization and excess deposition, mainly in cartilaginous tissue, mucous membranes, skin, bone surface, and cardiac structures. In addition, HA is excreted in biological solutions (urine, sweat, and semen) [2,5]. Therefore, the clinical presentation of alkaptonuria is quite varied and requires a systemic assessment of patients.
The main characteristics of endogenous ochronosis are ochronotic osteoarthropathy, aciduria, and deposition of melanin-like pigment [1,2].
Skin changes occur in photo-exposed areas and areas with apocrine glands. The most common sites of pigment deposition are the pinna and the ears. In the case described above, we present the involvement of the nail bed, which is rarely reported in the literature [2].
Ochronotic osteoarthropathy mainly affects the axial skeleton and joints under high pressure and can become progressively more severe [1,2,5].
Other systemic features include stone formation, osteopenia, fractures, tendon, muscle, and ligament ruptures, impairment of the pulmonary system, hearing loss, and valvular heart disease [4]. The main complications are valve calcifications and osteoarthritis [6].
There are few reports of patients with pigment deposits on the tympanic membrane and hearing loss, predominantly of a mixed pattern. The conduction component is inferred to be explained by the ochronotic change in the elasticity of the tympanic membrane or by changes in ossicular mobility [5].
The gold standard for diagnosis is the presence of high levels of urinary HA. In addition, the histopathological examination of different tissues can help with the diagnosis [3].
Although patients do not have an altered life expectancy, ochronotic osteoarthropathy causes significant morbidity. The basis of treatment is the control of joint symptoms with analgesia, mainly non-steroidal anti-inflammatory drugs, physical activity, and physical therapy. In addition, nitisinone, a potent inhibitor of 4-hydroxyphenylpyruvate dioxygenase, works by decreasing the production and urinary excretion of homogentisic acid, which is an important support for controlling the disease [7].
The follow-up of these patients requires a multidisciplinary approach, including dermatological, cardiological, orthopedic, ophthalmological, otorhinolaryngological, physiotherapy, and nutritional assessments.
Conflict of Interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Informed consent was obtained for this publication.
Keywords
Alkaptonuria; Metabolic disease; Ochronotic osteoarthropathy
Cite this article
Maciel RF, Boza JC, Maciel RF, Lira FE, Silveira LL, Oliveira MP. Alkaptonuria: Case report. Clin Case Rep J. 2025;6(1):1–4.
Copyright
© 2025 Rochelle Figini Maciel. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
