Long-Term Disease Stability in Essential Thrombocythemia with Double Driver MPL and CALR Mutations: A Case Report
* Sanghe J;
Wildman EC;
Chin-Yee B;
Hsia CC;
-
* Sanghe J: Schulich School of Medicine, Western University, London, Canada
-
Wildman EC: Department of Medicine, Western University, London Health Sciences Centre, Canada
-
Chin-Yee B: Department of Pathology and Laboratory Medicine, Western University, London Health Sciences Centre, Canada; Department of Medicine, Division of Hematology, Western University, London Health Sciences Centre, Canada
-
Hsia CC: Department of Pathology and Laboratory Medicine, Western University, London Health Sciences Centre, Canada; Department of Medicine, Division of Hematology, Western University, London Health Sciences Centre, Canada
Abstract
Myeloproliferative neoplasms (MPNs) are a group of hematologic disorders that are primarily associated with driver mutations in the Janus kinase 2 (JAK2), Calreticulin (CALR), and Thrombopoietin Receptor (MPL) genes. While these mutations were typically thought to be mutually exclusive, recent studies have reported particularly poor prognosis in cases with double driver mutations. We report the case of an 85-year-old woman diagnosed with Essential thrombocythemia (ET) over 30 years ago, with confirmed double driver MPL and CALR type 2 mutations. This patient has maintained a stable disease course on hydroxyurea, with no major adverse clinical events noted. This case report highlights the clinical variability and unpredictability that can occur in patients with double driver mutations and further emphasizes the ongoing need for additional research to better understand the complex interactions between double driver mutations and their impact on disease progression, treatment response, and prognosis.
Abbreviations
MPNs: Myeloproliferative neoplasms;
JAK2: Janus kinase 2;
CALR: Calreticulin;
MPL: Thrombopoietin receptor;
ET: Essential thrombocythemia;
PMF: Primary myelofibrosis;
PV: Polycythemia vera
Introduction
Philadelphia-negative myeloproliferative neoplasms (MPNs) are a heterogeneous group of hematologic malignancies that include Essential thrombocythemia (ET), Polycythemia vera (PV), and Primary myelofibrosis (PMF). A hallmark of MPNs is the presence of driver mutations in the Janus kinase 2 (JAK2), Calreticulin (CALR), and Thrombopoietin receptor (MPL) genes [1,2]. Driver mutations in JAK2, CALR, and MPL lead to constitutive activation of the JAK2 signal transducer and activator of transcription 5 (STAT5) pathway, resulting in abnormal proliferation of myeloid precursor cells and dysregulated production of erythrocytes, leukocytes, or platelets [3]. MPNs are typically prognosticated using the International prognostic score for thrombosis in Essential thrombocythemia (IPSET) score, which assesses the risk of thrombosis based on factors including age, history of thrombosis, cardiovascular risk, and platelet count. It categorizes patients into low, intermediate, and high-risk groups for thrombotic events [4]. Management of ET often requires cytoreductive therapy for patients at higher risk of thrombosis, and hydroxyurea is typically considered first-line [5].
CALR mutations in MPNs are predominantly gain-of-function frameshift mutations occurring in exon 9. Over 80% belong to one of two distinct mutation types [6]. The CALR type 1 mutation is a 52-bp deletion. The type 2 mutation is a 5-bp TTGTC insertion [6]. A previous study noted significantly higher platelet counts in individuals with a CALR type 2 mutation [7]. Notably, despite a higher platelet count, type 2 CALR mutations are associated with a lower risk of thrombosis and a more indolent clinical course [8].
It was initially thought that driver mutations in JAK2, CALR, and MPL occurred mutually exclusively in patients with MPNs. However, recent literature describes the co-occurrence of these mutations [9,10]. Diagnosis of an MPN with double driver mutations occurs in approximately 0.5% of patients, typically in older individuals with elevated platelet counts compared to those with single mutations [10]. Due to the rarity of this clinical entity, little information is otherwise known regarding the long-term clinical outcomes of patients with MPNs and double driver mutations.
We present a case of a patient with ET diagnosed over 30 years ago, with both MPL and CALR type 2 mutations, who has had a stable disease course with no major adverse events. This case adds valuable information to the literature, given the long-standing follow-up period in a patient with double-driver mutations. Additionally, we provide further data regarding the clinical course of a patient with a CALR type 2 mutation.
Case Presentation
An 85-year-old female currently followed in our Hematology clinics was initially diagnosed with ET in 1987 after admission to the hospital for a radical mastectomy for a new diagnosis of breast cancer. She was noted to have incidental thrombocytosis, which ultimately led to a clinical diagnosis of ET. At the time of referral to our clinic, her comorbidities included hypertension, hyperthyroidism, gastroesophageal reflux disease, and breast cancer. During her follow-up, she developed dementia, macular degeneration, squamous cell carcinoma, depression, and anxiety. The earliest laboratory values noted in our system were in 2003, at which time her hemoglobin was 133 g/L, MCV was 87 fL, RDW was 15.4 L/L, and platelet count was 979 x 10⁹/L, prior to initiation of cytoreductive therapy. Other diagnostic results include normal ferritin levels ranging from 46 μg/L to 174 μg/L, from 2010 to present. Peripheral blood films from 2008 to present have shown isolated moderately increased platelets without other abnormalities. In 2011, Polymerase Chain Reaction (PCR) testing for the typical JAK2 V617F mutation was negative but subsequent myeloid Next Generation Sequencing (NGS) panel revealed type 2 CALR: c.1154_1155insTTGTC, p.(Lys385Asnfs*47) (49%) and MPL: c.1771T>G, p.(Tyr591Asp) (44.5%) mutations.
Due to initial concerns about side effects, the patient had refused treatment until 16 years after diagnosis, when she started hydroxyurea 500 mg orally twice daily. Since the initiation of treatment, the patient has remained stable without any adverse events or need for therapies beyond hydroxyurea and aspirin. She has had hydroxyurea dose adjustments over the years to maintain her platelet counts below a target of 600 × 10⁹/L, but has been clinically well on a daily oral dose of 1000 mg since 2021. She was briefly on aspirin 81 mg orally daily in 2010, but she discontinued it due to epistaxis. It was reintroduced in 2023. She has also had brief periods off hydroxyurea, again due to concerns about side effects (potential hair loss) and social stressors. Her platelet counts have varied over the years, with levels above 600 × 10⁹/L, but hydroxyurea uptitration has been limited due to patient reluctance and macrocytic anemia. Her platelet counts over time are outlined in Figure 1. Despite varying platelet counts and periods without treatment, the patient has not experienced any thrombotic or other adverse clinical events secondary to ET.
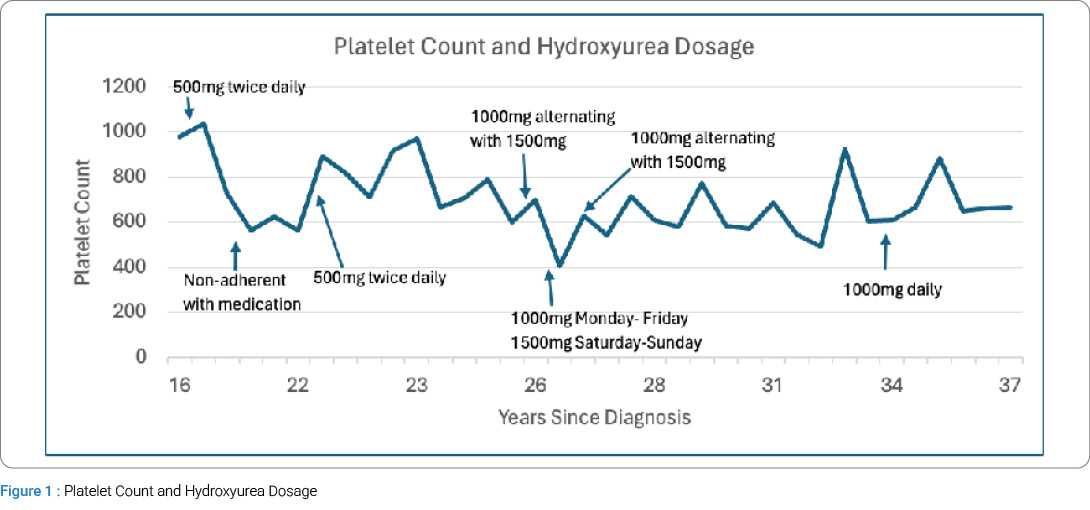
Discussion
This case report offers insight into the clinical phenotype of ET patients with double driver mutations and highlights differences between CALR type 1 and type 2 mutations. We follow an 85-year-old female with ET, carrying MPL and CALR type 2 mutations, to examine her decades-long, stable disease progression with hydroxyurea, despite intermittent adherence.
Building on these observations, studies comparing clinical outcomes between patients with CALR type 1 versus type 2 mutations have reported variable results. Type 1 mutations, involving a 52-bp deletion at the C-terminal domain, are generally associated with a more favorable prognosis, such as lower risk of thrombosis and better overall survival, whereas type 2 mutations, defined by a 5-bp insertion, tend to be linked to higher platelet counts and may be associated with different clinical features. Functionally, mutant CALR binds to the thrombopoietin receptor MPL and facilitates abnormal activation of JAK2 and STAT5 signaling, leading to uncontrolled megakaryocyte proliferation [11].
A 2014 study of 51 CALR type 1 and 44 CALR type 2 patients reported higher platelet counts in type 2 despite similar IPSET scores [7]. Larger studies show type 1 mutations more often lead to myelofibrosis and higher thrombotic risk [8,12,13]. Our patient has had a lower platelet count over time compared to median values for CALR type 2 in those studies, yet a more indolent disease course, aligning with these findings. Direct comparisons remain challenging due to the presence of an additional driver mutation. These findings highlight the need to further explore how specific mutations influence patient outcomes.
The Table 1 summarizes previous case reports that followed patients diagnosed with ET and double driver mutations. Our literature search to identify case reports of patients with ET and double driver mutations revealed only one case of a patient with MPL and CALR type 2 mutations [14]. The case was a 67-year-old female with MPL and CALR type 2 mutations who experienced transformation of her disease 3.5 months after diagnosis, and death within 2 years, despite aggressive treatment [14]. As the disease progressed, the CALR type 2 mutation was no longer present, suggesting that the MPL mutation may have been the more dominant clone. These findings contrast our patient with the same double driver mutations, who has remained stable for over 30 years.
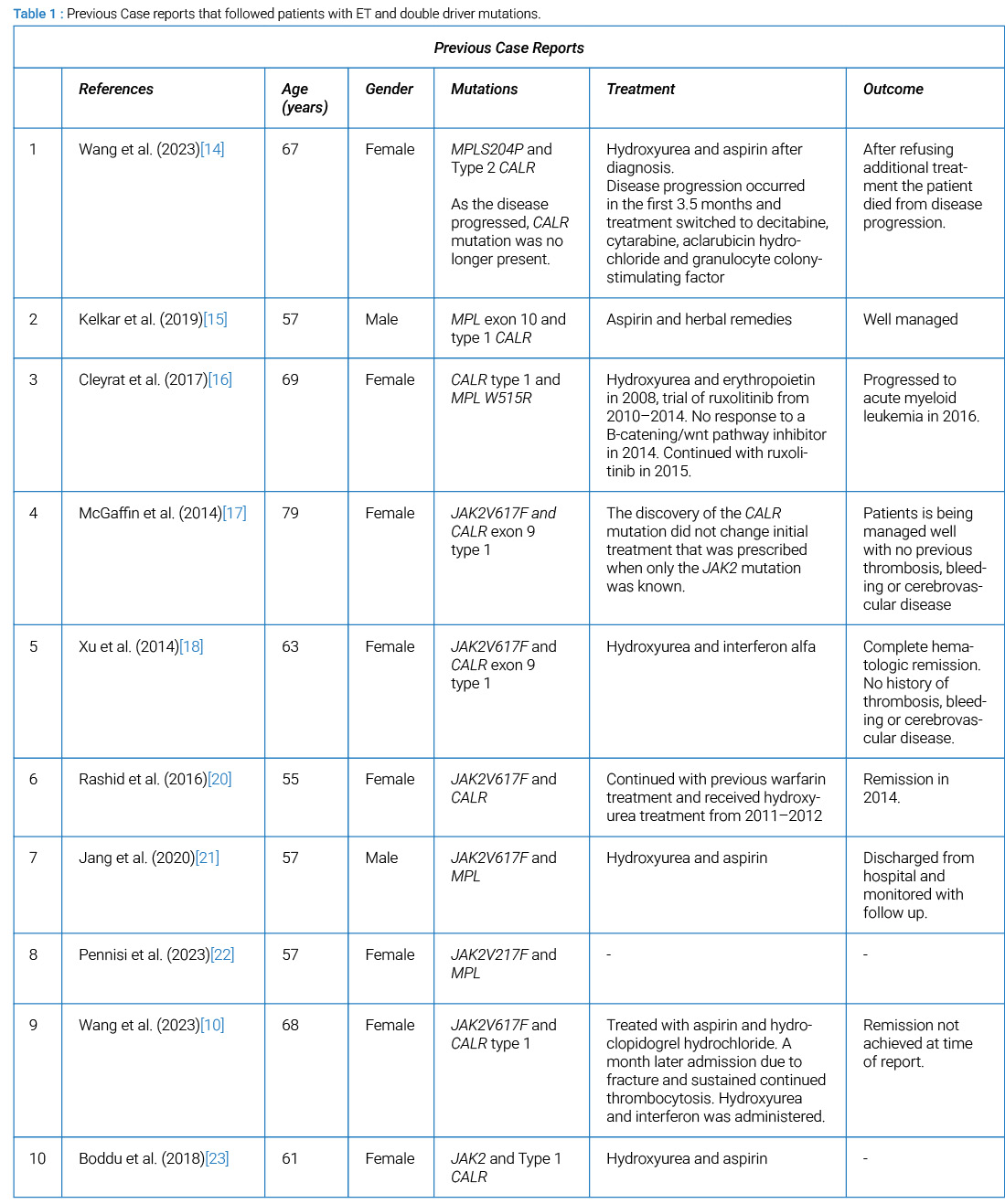
A previous case report discusses a 57-year-old male with MPL and CALR type 1, who has been well managed on aspirin and herbal remedies [15]. Another report of a 69-year-old female with the same driver mutations, who faced disease progression into acute myeloid leukemia, despite undergoing various treatments [16]. This difference in disease progression may be attributed to additional mutations or clonal evolution, highlighting the variability that can occur. Other studies on patients with JAK2 V617F and CALR exon 9 type 1 mutations showed comparable disease progression, with no thrombotic nor hemorrhagic events noted [17,18]. Theoretical mechanisms for the differences in disease progression in patients with double driver mutations may involve the presence of two distinct clones, each harboring a different mutation. Such clonal coexistence could allow for independent contributions to pathogenesis, leading to complex disease behavior and varied treatment responses [19]. The dominant mutation may significantly influence clinical outcomes, underscoring the importance of identifying co-occurring mutations for prognosis and treatment planning.
Limitations to this case study include our lack of access to the patient’s initial laboratory results and presenting symptoms, both of which would have enhanced our understanding of her disease progression. Additionally, the lack of a bone marrow biopsy limits the comprehensiveness of our evaluation. It is not known whether the patient had both driver mutations present at the time of diagnosis, more than 37 years ago, as molecular diagnostic tests for JAK2 V617F and myeloid Next Generation Sequencing (NGS) were not available at our institution until 2007 and 2018, respectively. Thus, we cannot determine which driver mutations occurred first or which represents the dominant clone. Further, single-cell sorting techniques are not available to us to distinguish between the presence of a single clone with both mutations versus two distinct clones, each with its own mutation.
In summary, this case report provides valuable insights into the clinical variability of ET in patients with double driver mutations, particularly MPL and CALR type 2 mutations. The patient’s stable disease course over three decades contrasts with poorer clinical outcomes reported in similar cases, emphasizing the heterogeneity of disease progression. These findings highlight the importance of considering the interplay between multiple mutations and the potential for individualized disease trajectories, underscoring the need for further research into the mechanisms driving these differences and their implications for treatment strategies.
Acknowledgments
No financial support or funding was received for the preparation of this case report.
Authors’ Contribution
Conceptualization: Jessie Sanghe, Emily Wildman, Cyrus Hsia; Data curation: Jessie Sanghe, Emily Wildman, Cyrus Hsia,
Analysis: Jessie Sanghe, Emily Wildman, Cyrus Hsia;
Project administration: Cyrus Hsia;
Resources: Cyrus Hsia;
Visualization: Jessie Sanghe;
Writing- original draft: Jessie Sanghe;
Writing- review & editing: Emily Wildman, Benjamin Chin-Yee, Cyrus Hsia;
Senior authors: Cyrus Hsia, Benjamin Chin-Yee
Ethical Conduct Approval – Helsinki – IACUC: As per institutional guidelines from London Health Sciences Center, ethics approval was not required for the publication of this case report. Informed consent was obtained from the patient for publication.
Informed Consent Statement: All authors and institutions have confirmed this manuscript for publication.
Data Availability Statement: All are available upon reasonable request.
Competing of Interest
No competing interests were disclosed.
References
- Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36(7):1703–1719.
- Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. International consensus classification of myeloid neoplasms and acute leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140:1200–1228.
- Greenfield G, Mc Mullin MF, Mills K. Molecular pathogenesis of the myeloproliferative neoplasms. J Hematol Oncol. 2021;14(1):103.
- Alvarez-Larrán A, Cuevas B, Velez P, Noya S, Caballero-Navarro G, Ferrer-Marín F, et al. Application of IPSET-thrombosis in 1366 patients prospectively followed from the Spanish registry of essential thrombocythemia. Hemasphere. 2023;7(8):e936.
- Harrison CN, Campbell PJ, Buck G, Wheatley K, East CL, Bareford D, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med. 2005;353(1):33–45.
- Imai M, Araki M, Komatsu N. Somatic mutations of calreticulin in myeloproliferative neoplasms. Int J Hematol. 2017;105(6):743–747.
- Tefferi A, Wassie EA, Guglielmelli P, Gangat N, Belachew AA, Lasho TL, et al. Type 1 versus type 2 calreticulin mutations in essential thrombocythemia: A collaborative study of 1027 patients. Am J Hematol. 2014;89(8):E121–E124.
- Pietra D, Rumi E, Ferretti VV, Buduo CADi, Milanesi C, Cavalloni C, et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia. 2016;30(2):431–438.
- De Roeck L, Michaux L, Debackere K, Lierman E, Vandenberghe P, Devos T. Coexisting driver mutations in MPN: Clinical and molecular characteristics of a series of 11 patients. Hematology. 2018;23(10):785–792.
- Wang Y, Ran F, Lin J, Zhang J, Ma D. Genetic and clinical characteristics of patients with Philadelphia-negative myeloproliferative neoplasm carrying concurrent mutations in JAK2V617F, CALR, and MPL. Technol Cancer Res Treat. 2023;22:15330338231154092.
- Iborra FJ, Papadopoulos P. Calreticulin in essential thrombocythemia: Stressing out the megakaryocyte nucleus. Front Oncol. 2017;7:103.
- Pérez Encinas MM, Sobas M, Gómez-Casares MT, Abuin Blanco A, Noya Pereira MS, Raya JM, et al. The risk of thrombosis in essential thrombocythemia is associated with the type of CALR mutation: A multicentre collaborative study. Eur J Haematol. 2021;106(3):371–379.
- Loscocco GG, Guglielmelli P, Gangat N, Rossi E, Mannarelli C, Betti S, et al. Clinical and molecular predictors of fibrotic progression in essential thrombocythemia: A multicenter study involving 1607 patients. Am J Hematol. 2021;96(11):1472–1480.
- Wang J, Fu W, Bao W, Gong W, Xu S, Ling C, et al. Genomics of clonal evolution in a rare essential thrombocythemia with coexisting type 2 CALR and MPL S204P mutations. Platelets. 2023;34(1):2176167.
- Kelkar K, Ramanan V, Anand S, Ranade S, Patil K, Agarwal M, et al. Co-occurrence of CALR and MPL somatic mutations in an Indian patient with a Philadelphia-negative myeloproliferative neoplasm. J Hematopathol. 2019;12:163–168.
- Cleyrat C, Chabot-Richards DS, Lynch DT, Gotlib J, George TI, Wilson BS, et al. Leukemic transformation of post-essential thrombocythemia myelofibrosis: A unique case presenting with double MPL and CALR mutations. Blood. 2017;130:4215.
- McGaffin G, Harper K, Stirling D, McLintock L. JAK2 V617F and CALR mutations are not mutually exclusive; findings from retrospective analysis of a small patient cohort. Br J Haematol. 2014;167(2):276–278.
- Xu N, Ding L, Yin C, Zhou X, Li L, Li Y, et al. A report on the co-occurrence of JAK2V617F and CALR mutations in myeloproliferative neoplasm patients. Ann Hematol. 2015;94(5):865–867.
- Thompson ER, Nguyen T, Kankanige Y, Yeh P, Ingbritsen M, McBean M, et al. Clonal independence of JAK2 and CALR or MPL mutations in comutated myeloproliferative neoplasms demonstrated by single cell DNA sequencing. Haematologica. 2021;106(1):313–315.
- Rashid M, Ahmed RZ, Ahmed S, Nadeem M, Ahmed N, Shamsi TS. Coexisting JAK2V617F and CALR exon 9 mutation in essential thrombocythemia. Indian J Hematol Blood Transfus. 2016;32(Suppl 1):112–116.
- Jang MA, Seo MY, Choi KJ, Hong DS. A rare case of essential thrombocythemia with coexisting JAK2 and MPL driver mutations. J Korean Med Sci. 2020;35(23):e168.
- Pennisi MS, Di Gregorio S, Tirrò E, Romano C, Duminuco A, Garibaldi B, et al. Additional genetic alterations and clonal evolution of MPNs with double mutations on the MPL gene: Two case reports. Hematol Rep. 2023;15(2):317–324.
- Boddu P, Chihara D, Masarova L, Pemmaraju N, Patel KP, Verstovsek S. The co-occurrence of driver mutations in chronic myeloproliferative neoplasms. Ann Hematol. 2018;97(11):2071–2080.
Keywords
Essential thrombocythemia; Myeloproliferative neoplasms; CALR type 2 mutation; MPL mutation
Cite this article
Sanghe J, Wildman EC, Chin-Yee B, Hsia CC. Long-Term Disease Stability in Essential Thrombocythemia with Double Driver MPL and CALR Mutations: A Case Report. Clin Case Rep J. 2026;7(2):1–5.
Copyright
© 2026 Sanghe J. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
