Overlap of Behcet’s Disease and Systemic Sclerosis: A Case Report
Somayeh Soroureddin;
Ali Baradaran Bagheri;
Seyedeh Zahra Badrkhahan;
Negar Movasseghi Jourshari;
* Majid Alikhani;
-
Somayeh Soroureddin: Rheumatology Fellow, Department of Internal Medicine, Rheumatology Research Center, Shariati Hospital, Tehran.
-
Ali Baradaran Bagheri: Assistant Professor, Department of Neurosurgery, Shahid Madani Hospital, Alborz University of Medical Science, Karaj, Iran.
-
Seyedeh Zahra Badrkhahan: Geriatric Medicine Fellow, Department of Geriatric Medicine, Tehran University of Medical Sciences, Tehran, Iran.
-
Negar Movasseghi Jourshari: Rheumatology Research Center, Shariati Hospital, Tehran University of Medical Science, Tehran, Iran.
-
* Majid Alikhani: Assistant Professor, Department of Internal Medicine, Rheumatology Research Center, Shariati Hospital, Tehran University of Medical Science, Tehran, Iran.
Abstract
Background: Behcet’s Disease (BD) and Systemic Sclerosis (SSc) are two distinct rheumatic disorders with few overlapping clinical manifestations. This case report highlights the co-occurrence of BD and SSc in a 43-year-old female patient and discusses the challenges in diagnosis and management.
Case presentation: The patient initially presented with features consistent with BD, including oral aphthosis and ocular lesions. Treatment with immunosuppressive therapy resulted in disease remission. However, several years later, the patient developed sclerotic skin changes, dyspnea, and dysphagia, fulfilling the criteria for SSc. Treatment strategies were tailored based on the manifestations of both diseases.
Discussion: The overlap of BD and SSc poses unique challenges in diagnosis and treatment. Genetic and environmental factors play a role in the pathogenesis of both diseases. The presence of potential shared genetic susceptibility loci may contribute to their coexistence. This case report underscores the complexity of autoimmune diseases and highlights the need for a comprehensive and multidisciplinary approach to managing patients with coexisting BD and SSc. The overlapping features and distinct manifestations of these diseases necessitate tailored treatment strategies. Further research is required to understand better the underlying mechanisms driving their coexistence and to establish optimal management approaches for such patients.
Conclusion: The key findings indicate that both genetic and environmental factors may contribute to the coexistence of BD and SSc, suggesting potential shared susceptibility loci. Clinically, the necessity for a tailored, multidisciplinary treatment approach becomes evident due to these diseases’ distinct and overlapping manifestations. This case highlights the importance of vigilant long-term monitoring and adaptable treatment strategies for clinical practice.
Introduction
Behcet’s Disease (BD) and Systemic Sclerosis (SSc) are two distinct rheumatic disorders characterized by systemic inflammation and involvement of multiple organ systems. Although these diseases have unique clinical manifestations, there is limited evidence regarding their co-occurrence, suggesting a potential interplay between their pathogenic mechanisms.
BD is a chronic, relapsing systemic vasculitis that primarily affects young adults, with a higher prevalence in countries along the ancient Silk Road, including Turkey, Iran, and Japan. It is characterized by recurrent oral and genital ulcers, ocular inflammation (uveitis), skin lesions, and involvement of various organ systems. The exact etiology of BD remains elusive; however, it is believed to involve complex interactions between genetic predisposition, environmental factors, and dysregulated immune responses. The HLA-B51 allele has been strongly associated with BD susceptibility, suggesting a genetic component. Additionally, infectious triggers, such as Streptococcus sanguinis and herpes simplex virus, have been proposed as potential initiators of the immune response in genetically susceptible individuals.
Systemic Sclerosis (SSc), also known as scleroderma, is an autoimmune connective tissue disorder characterized by fibrosis of the skin and internal organs. It predominantly affects women in their middle age, with a higher incidence in individuals of African descent. Excessive collagen deposition, vascular abnormalities, and immune dysregulation characterize SSc. The pathogenesis of SSc also involves complex interactions between genetic, environmental, and immunological factors. Genetic studies have identified several susceptibility genes, including HLA alleles, TGF-β signaling pathway genes, and genes involved in the endothelin-1 pathway. Environmental triggers such as silica exposure, organic solvents, and certain medications have also been implicated in disease development.
Despite their distinct clinical features, BD and SSc share some commonalities in their pathogenic mechanisms. In BD, immune dysregulation primarily affects the vascular endothelium, leading to endothelial dysfunction and subsequent vasculitis. In SSc, immune dysregulation is characterized by an exaggerated immune response, activation of fibroblasts, and excessive collagen deposition, resulting in tissue fibrosis. Furthermore, aberrant angiogenesis and vascular abnormalities are observed in both diseases [1], contributing to their clinical manifestations. However, notable differences exist in the pathogenesis of BD and SSc. BD is characterized by a predominantly Th1 and Th17 immune response, with elevated levels of pro-inflammatory cytokines such as interferon-gamma (IFN-γ) and interleukin-17 (IL-17). In contrast, SSc is associated with a Th2 skewed immune response, with increased production of profibrotic cytokines such as transforming growth factor-beta (TGF-β) and interleukin-4 (IL-4). Moreover, the involvement of specific signaling pathways, such as the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway in BD and the TGF-β pathway in SSc, further differentiates their pathogenic mechanisms.
As such, reporting the co-occurrence of these diseases, which are less likely to be expected from a pathological point of view, provides further insight to the pathogenesis of both diseases. Herein, we report a long-term case of BD with adequate disease control that was subsequently complicated with symptoms of SSc.
Case Presentation
The patient presented is a 43-year-old female with a known case of BD since 15 years ago. She was previously diagnosed with The International Criteria for Behcet’s Disease (ICBD) based on the presentation of oral aphthosis (2 points), ocular lesions (2 points), and erythema nodusom (1 point). The ocular lesions consisted of pan-uveitis and macular edema. Previous workups revealed that the patient also harbors Human Leukocyte Antigen-B5 (HLA-B5) and HLA-B51. Due to the pronounced presentation of BD and pan-uveitis, the patient was initially receiving immunosuppressive therapy with Cyclophosphamide and high-dose Prednisolone, as well as Azathioprine (2.5 mg/kg), and 1 mg colchicine. Subsequent unremarkable follow-up visits and adequate disease control led to gradually tapering the administered agents.
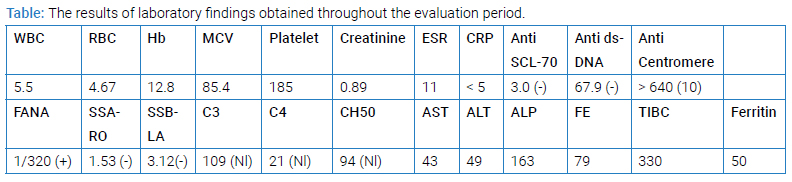
Around five years ago, the patient first noticed of discoloration of the fingers, a pattern compatible with Raynaud’s phenomenon. No other symptoms and signs were compatible with other autoimmune diseases, like connective tissue disease and vasculitis. Additional workups, including serology tests, were requested. BD was in remission at that time with 5 mg daily prednisone and 1 mg daily colchicine. Lab results reported an Antinuclear Antibody (ANA) level of 5.6 IU (Normal <1.1 IU), Anti-Centromere antibody level of >200 AU/mL (Normal: <20 AU/mL), and Anti-Scl-70 antibody level of < 2 AU/mL (Normal: <2 0 AU/mL). Other serologic tests in favor of connective tissue disease and vasculitis were negative. Capillaroscopy showed an early scleroderma pattern, which means the presence of few enlarged/giant capillaries, few capillary hemorrhages, and no evident loss or distortion of the capillaries. No signs of sclerosis and skin changes were observed at that time. Based on clinical presentation (Raynaud’s phenomenon), laboratory findings (FANA and Anti-Centromere positive test), and capillaroscopy, the patient was started on an additional treatment course with daily Aspirin and 60 mg Diltiazem.
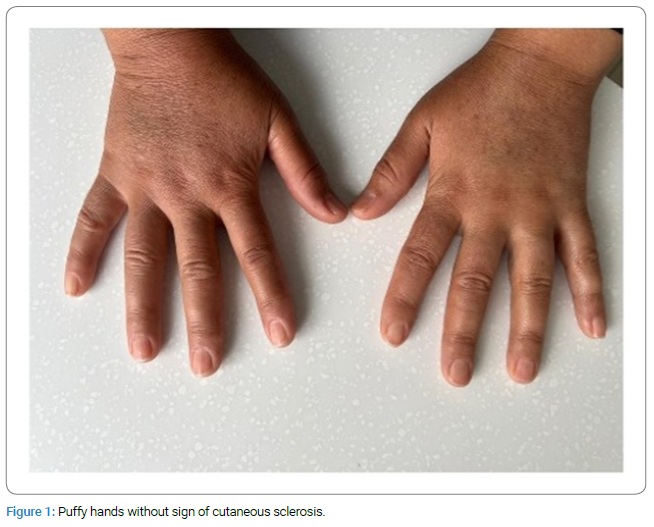
The patient was lost to follow-up until six months ago when they presented with exacerbated Raynaud’s phenomenon, puffy hands (Figure 1), grade 2 dyspnea according to the modified Medical Research Council Dyspnea Scale (mMRC), and dysphagia that had been ongoing for the past 7 months–8 months. Repeated testing revealed FANA results to be positive with a > 1/320 (1/40) titer and Anti-Centromere Ab > 640 (> 10). Pulmonary function testing with spirometry was unremarkable. High-Resolution Computed Tomography (HRCT) scan of the lungs was normal (Figure 2). The endoscopy did not show any signs in favor of scleroderma involvement. Echocardiography did not reveal any specific findings and was not suggestive of pulmonary hypertension. A subsequent capillaroscopy showed giant capillaries, enlarged loops, and microbleeding. Ophthalmic physical examination to evaluate BD activity was suggestive of adequate disease control. At the time, the patient fulfilled EULAR/ACR 2013 criteria for systemic sclerosis (Reynaud’s phenomenon = 3, Puffy fingers = 2, Anti-Centromere = 3, Abnormal capillaroscopy = 2). Thus, Prednisolone increased to 7.5 mg, Methotrexate 15 mg, ASA, and 60 mg Diltiazem were prescribed. The patient was diagnosed with an overlap syndrome of BD and systemic sclerosis. The follow-up visit six months later was uneventful, with disease remission and symptom control. A summary of laboratory findings throughout the evaluation period is given in (Table).
Discussion
The co-occurrence of BD and systemic sclerosis in this patient is a rare finding. The development of systemic sclerosis in a patient with a known history of BD raises questions about the underlying pathogenic mechanisms and the potential interplay between these two autoimmune disorders. While the precise relationship between these conditions remains unclear, there have been reports of similar cases indicating a possible association [2–4]. Watanabe et al. summarize these cases’ characteristics [4]. The treatment in the case reported by Yokota et al. consisted of 5 mg/day–30 mg/day methylprednisolone for the initial scleroderma diagnosis, which was increased to 30 mg upon the diagnosis of the entero-Behcet’s Disease [5]. For Sjogren’s syndrome complicated by SSc and then BD in the patient reported by Watanabe et al., prednisolone was given 15 mg initially and then tapered and replaced by colchicine.
One of the challenges in managing such patients is determining the optimal treatment course after the onset of both conditions. In this case, the initial focus was on controlling the manifestations of BD, particularly the ocular involvement, with immunosuppressive therapy. However, the subsequent development of systemic sclerosis necessitated reevaluating the treatment approach. The presence of the Raynaud phenomenon, dyspnea, and dysphagia indicates systemic involvement and highlights the need for comprehensive management.
Treatment strategies for systemic sclerosis aim to alleviate symptoms, prevent organ damage, and improve quality of life. Multidisciplinary care involving rheumatologists, dermatologists, and other specialists is crucial in providing comprehensive care for these patients. Depending on the extent and severity of organ involvement, the treatment plan may include immunosuppressive agents, vasodilators, and medications targeting fibrosis.
In conclusion, the overlap of BD and systemic sclerosis in this patient presents a unique clinical challenge. These two autoimmune disorders’ overlapping features and distinct manifestations require careful management and close monitoring. Further research is needed to elucidate the underlying mechanisms driving their coexistence and to establish optimal treatment approaches for such patients.
Acknowledgments
Limitation
Our limitation was not having access to special autoantibodies like RNA polymerase.
Ethical Approval
Written informed consent to publish this case report and the accompanying clinical data and images were obtained from the patient prior to the preparation of this manuscript. The case report adhered to was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments.
Funding
The authors did not receive support from any organization for the submitted work.
Author Contributions
Somayeh Soroureddin was the primary physician of the patient and had a principal role in data collection.
Data Availability Statement
Complete clinical and imaging data of the patient is available from the corresponding author on reasonable request.
Conflict of Interest
All authors state that no financial and personal relationships with other people or organizations that could inappropriately influence (bias) their work.
References
- Maruotti N, Cantatore FP, Nico B, Vacca A, Ribatti D. Angiogenesis in vasculitides. Clinical & Experimental Rheumatology. 2008;26(3):476–483.
- Choy E, Kingsley G, Panayi G. Systemic sclerosis occurring in a patient with Adamantiades-Behcet’s disease. Br J Rheumatol. 1993;32(2):160–161.
- Muroya T, Ishii Y, Chisaka R. [Progressive systemic scleroderma suspected to be Behcet’s disease]. Hokkaido Igaku Zasshi. 1974;49(6):567–568.
- Watanabe H, Yashiro M, Asano T, Sato S, Takahashi A, Katakura K, et al. A case of behcet’s disease and systemic sclerosis developing after an earthquake disaster. Fukushima J Med Sci. 2015;61(1):86–90.
- Yokota K, Hirano M, Akiba H, Adachi D, Takeishi M, Akiyama Y, et al. [A case of Behcet’s disease with esophageal ulcers complicated with systemic sclerosis, chronic hepatitis C, and pancytopenia]. Nihon Rinsho Meneki Gakkai Kaishi. 2004;27(3):164–170.
Keywords
Behcet’s disease; Systemic sclerosis; Overlap
Cite this article
Soroureddin S, Bagheri AB, Badrkhahan SZ, Jourshari NM, Alikhani M. Overlap of behcet’s disease and systemic sclerosis: a case report. Clin Case Rep J. 2024;5(1):1–4.
Copyright
© 2024 Majid Alikhani. This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY-4.0).
